
Abstract
Monozygotic twin brothers with a subtelomeric 6q deletion presented with mental retardation, microcephaly, seizures, an enlarged cisterna magna, dimpling at elbows, a high arched palate and a thin upper lip. The same subtelomeric deletion was detected in the mother of the patients, presenting with a milder phenotype. We narrowed down the breakpoint to a region of approximately 100 kb and estimated the size of the terminal deletion to be 1.2 Mb. This region contains four known and seven putative genes. Comparison of the deletion with other reported patients showed TBP was the most plausible candidate gene for the mental retardation in this syndrome. We verified that the TBP gene expression was halved in our patients using real-time PCR. Cognitive and behavioural tests performed on previously described heterozygous tbp mice suggested that TBP is potentially involved in cognitive development.
Similar content being viewed by others


Introduction
Subtelomeric rearrangements account for approximately 5–7% of all mental retardation cases.1, 2, 3 Apart from a few chromosome ends that cause recognizable syndromes such as Wolf–Hirschhorn syndrome (caused by a deletion of the tip of chromosome 4p) or Miller–Dieker syndrome (caused by a deletion of chromosome 17p), for only some of the other deleted chromosome ends, a genotype–phenotype correlation has been postulated, including subtelomeric 1p and 1q deletions.4, 5 Recently, a new syndrome was recognized in patients with a 9q subtelomeric deletion.6 The Eu-HMTase gene was subsequently identified as the causative gene for mental retardation in these aberrations.7 For many subtelomeric deletions, too few cases have been reported to allow a definition of a phenotype.
Subtelomeric rearrangements involving chromosome 6q have been reported in a limited number of studies and the size of the deletion varied from less than 0.5 Mb to over 8 Mb.8 Frequently observed symptoms include developmental delay, seizures, hypoplasia of the corpus callosum, dimpling on elbows and knees and specific dysmorphic features.9 Patients carrying a subtelomeric 6q deletion share phenotypic features with patients with cytogenetically visible 6q deletions. This may imply that genes responsible for the phenotype are located in the most distal part of the 6q chromosome. Therefore, it is important to study the extent of these genomic imbalances and the genes within those regions to refine the correlation between genotype and phenotype. Here, we report on monozygotic twin brothers carrying a small pure subtelomeric 6q deletion that was detected by MLPA previously.10 We fine mapped the breakpoint region and analysed the possible role of the TBP gene in this disorder.
Subjects, materials and methods
Case report
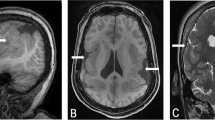
Monozygotic twin brothers were born at 35 weeks of gestation. They have moderate mental retardation, microcephaly (head circumference 50.8 and 53 cm at 18 years of age, <3rd percentile), hyperactivity and generalized epilepsy. They have epicanthal folds, a high arched palate, a thin upper lip and dimpling at the elbows. MRI revealed a larger cisterna magna and broadened occipital horns. The mother presented with a speech deficit, some dysmorphic features and had a lower than normal IQ.
Microsatellite marker analysis
Blood samples were obtained from the probands and their relatives. Genomic DNA was obtained from whole blood by salt extraction. PCR amplification of genomic DNA of parents and children was performed in a MJ research PTC 200 thermocycler and PCR products were subsequently analysed on a ABI 3100 capillary sequencer (Applied Biosystems, Foster City, CA, USA).
FISH analysis
Metaphase spreads were prepared from lymphocyte cultures using standard procedures. Probes were labelled by nick translation and were hybridized according to standard protocols. A dioxigenin-labelled probe was detected by an FITC-conjugated antibody α-dioxigenin detection system, and a biotin-labelled probe by an ALEXA-594-conjugated streptavidin detection system. Slides were counterstained with VECTA-SHIELD® containing 4′6-diamino-2-phenylindole (DAPI). Image capture and analyses were performed using a Leica fluorescence microscope equipped with the appropriate filter combination for detecting ALEXA-594, FITC and DAPI. The images were captured by a cooled CCD camera equipped with a Leica image analysis system.
Southern blotting experiments
Southern blotting experiments were performed according to standard protocols. Following a 4-h restriction endonuclease digestion, genomic DNA was separated by 0.7% agarose gel electrophoresis. Subsequently, the restriction fragments were transferred to a Nylon membrane (Hybond-N+, Amersham Biosciences, Piscataway, NJ) and were crosslinked by alkali. Probes of PCR-amplified genomic DNA were labelled with 32P-dATP and 32P-dCTP by Klenow reaction with random hexamer primers. Probes were hybridized in hybridization solution (10% Denhardts, 15% 20 × SSC, 4% SDS and 10% dextrane sulphate) with herring sperm DNA. Blots were washed with SSC/SDS wash solutions with elevating stringencies and exposed to a radiographic film. Primer pairs for amplification of the hybridization probes were based on the genomic sequence of contig NT_007302.12 and are as follows: PHF10 forward 5′-cactgcgcaatgctgttaagc-3′ and PHF10 reverse 5′-ttggtgaaatctgatactgtgag-3′.
Human multiple tissue expression (MTE) array
We used a human MTE array (BD Biosciences Clontech, Palo Alto, CA, USA) to study the expression pattern of the TBP gene. A cDNA probe specific for the TBP gene, located in the 3′UTR (forward 5′-AGATGGATGTTGAGTTGCAGG-3′ and reverse 5′ATATTCTGTCCTGCAATACTGG-3′) was designed and the assay was performed according to the manufacturer's recommendations.
Real-time PCR
RNA was isolated out of lymphocytes using a standard Trizol extraction. The quality of the RNA was tested on agarose gels and spectrophotometrically. Subsequently, cDNA was synthesized out of total RNA using the Superscript III First-strand synthesis system for RT-PCR (Invitrogen, Carlsbad, CA, USA). Next, the cDNA was treated with DNase I to remove potential genomic contaminations using a DNA-free kit (Ambion, Austin, TX, USA). Real-time PCR was performed on an ABI Prism 7000 sequence detector and a Taqman® gene expression assay specific for the TBP gene (assay ID Hs00427620_m1) was used (Applied Biosystems, Foster City, CA, USA). Three housekeeping genes were used for normalization: GAPD (assay ID Hs 99999905_m1), UBC (assay ID Hs00824723_m1) and YWHAZ (assay ID Hs00237047_m1).11 The cycling conditions comprise 2 min at 50°C, 10 s 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The assay was performed in duplo and included a no-template control for each gene. Data analysis were performed using the accompanied software (ABI Prism 7000 sequence detection system) and results were exported to Excel for further analysis. The relative gene expression levels were normalized to the geometric mean of the three selected house keeping genes.
Studies in mice
The mice used in our study were bred at the University of Strasbourg and transferred to the University of Leuven at the age of 2 months. The tbp+/− mice had been backcrossed onto a C57BL/6 background for nine generations, and wild-type littermates were used as controls. Animals were kept in mixed genotype groups of 4–6 males at standard laboratory conditions (12 h dark/light cycles, constant room temperature and humidity, standard lab chow and water ad libitum).
Neuromotor activity and exploratory behaviour
Grip strength Grip strength was measured using a device consisting of a T-shaped bar connected to a digital dynamometer (Ugo Basile, Comerio, Italy).12 Mice were placed before the bar, which they usually grabbed spontaneously, and gently pulled backwards until they released the bar (maximal readouts were recorded). Ten such measurements were obtained for each animal.
Rotarod Motor coordination and equilibrium was tested on an accelerating rotarod (Med Associates, St Albans, VT, USA).12 Mice were first trained at constant speed (4 r.p.m., 2 min) before they started with four test trials (intertrial interval 10 min). During these test trials, the animals had to balance on a rotating rod that accelerated from 4 to 40 r.p.m. in 5 min, and time until they dropped from the rod was recorded (up to 5 min cutoff).
Cage activity Cage activity was recorded using a lab-build activity logger connected to three infrared (IR) photobeams. Mice were put separately in 20 × 30 cm2 transparent cages, placed between the photo beams and activity was measured as beam crossings for each 30 min, during a 24 h interval.
Open field Open-field exploration was examined using a 50 × 50 cm2 square arena. Each animal was dark adapted for 30 min and placed in the arena for 10 min. Movements in the arena were recorded using Etho Vision video tracking equipment and software (Noldus, Wageningen, The Netherlands). Total path length and corner crossings were included as measures of locomotor activity. These measures are highly correlated and mainly indicate thigmotactic walking near the walls of the arena. Entries into the centre of the field were included as a measure of conflict resolution and anxiolysis.
Social exploration Social interaction is another measure of anxiety,13 where the amount of time spent was measured in the same open-field arena by placing two female mice in a centrally-located cage enabling visual, olfactory and limited physical contact with the tested male mouse. Mice started from a specific corner of the arena, and recording of the explorative pattern began after 1 min of adaptation. Total path lengths, as well as number of entries and path length in the centre circle during 10 min trial duration were recorded as measures of social exploration.
Anxiety and learning behaviour
Elevated plus maze The elevated plus maze was used as a test for anxiety-related exploration. The arena consisted of a plus-shaped maze with two arms (5 cm wide) closed by sidewalls and two arms without walls. Mice were placed at the centre of the maze and were allowed to explore freely for 10 min. Exploratory activity was recorded by five IR beams (four for arm entries and one for open arm dwell) connected to a computerized activity logger.
Passive avoidance Passive avoidance learning was assessed in two-compartment box with a shock grid.12 This task allows measurement of avoidance behaviour and learning abilities in mice. The box consisted of an illuminated compartment and a dark compartment, separated by a guillotine door. After 30 min adaptation to a dark environment, the subject was placed in the light part and latency to enter the dark compartment was measured. If the subject failed to enter the dark compartment within 5 min trial duration, it was gently probed to enter the dark part. Upon entry into the dark compartment, the door was closed and a 2 s foot shock (0.2 mA) was applied. The mouse was then removed from the box and placed in its home cage. After 24 h the dark-adapted mouse was again placed in the light box and latency to enter the dark compartment was measured.
Radial arm maze The radial arm maze allows one to test for working and spatial memory capabilities in a terrestrial environment. The dark grey PVC arena consisted of a central round platform (diameter 40 cm) from which eight enclosed arms (40 × 8 cm with a 5 cm wall) radiated. Manually operated guillotine doors allowed the closing of individual arms. At the end of each arm, a small food cup inserted in a hole contained the food bait. The maze was situated 80 cm above the floor and comprised extra-maze cues as well as different black-and-white signs posted at the arm entrances. A large cylinder made of black paper was placed on the platform at the start of the trial and the mouse was placed inside the cylinder. After 10 s, the trial was started by lifting the paper cylinder to allow access to the arms. After 5 days of handling and habituation to the maze (5 min/day), subjects were trained in standard radial maze tasks (eight choices) in one daily session. Each food cup contained a food bait (chocolate-covered rice crispies), and the subject was to obtain all food pellets within 10 min. The session ended when the subject either retrieved all eight food pellets or 10 min had passed. No visit or a repeat visit to an arm was counted as an error.
Statistics Data are presented as mean and SEM. Differences between groups were examined using unpaired two-tailed t-tests or ANOVA procedures with repeated measures. Significance levels were at P<0.05.
Results and discussion
The subtelomeric 6q deletion in the patients has been detected by MLPA and confirmed by FISH analysis using probe GS-57-H24 previously.10 The same probe was deleted in the mother, whereas no abnormalities were found in the father. The twins presented with a phenotype that resembles the 6q deletion phenotype including mental retardation, microcephaly, seizures, an enlarged cisterna magna, dimpling at elbows and dysmorphic features. Therefore, we assume this deletion is the cause of the clinical manifestations in the patients. The mother was mildly affected. Retrospective subtelomeric MLPA analysis was not indicative of mosaisim in the mother. Monosomy of the distal part of chromosome 6q has already been reported as the cause of very mild mental retardation in patients without dysmorphic features.14
Using microsatellite marker analysis (Table 1), we refined the localization of the breakpoint to the region between marker D6S446 (distance to telomere 730 kb, deleted) and D6S281 (distance to telomere 1384 kb, retained). Additional FISH analysis narrowed the breakpoint region further down to a region of approximately 150 kb between clone RP1-171-H20 (distance to telomere 1.3 Mb, retained) and clone RP1-182-D15 (distance to telomere 1.1 Mb, deleted). As no more known microsatellite markers or BAC clones were available in this 150 kb region, we performed Southern blotting on an XmnI digest of genomic DNA of the patients and controls with a probe for the PHF10 gene located in this region. This probe was not deleted in the patients, placing the breakpoint between clone RP1-182-D15 and the PHF10 gene, a region of approximately 100 kb (Figure 2).

Overview of the known genes in the deleted region. The breakpoint region of 100 kb is indicated. The smallest deletion region reported is indicated.
To determine whether the deletion arose de novo in the mildly affected mother (II-2), we also analysed the maternal grandmother (I-2) (the maternal grandfather (I-1) was not available). By haplotype analysis, we could determine that the deletion was likely not derived from the grandmaternal chromosome (I-2) (Figure 1). This means that the deletion was either inherited from the grandfather (I-1) or has arisen de novo (Figure 1).

Haplotype analysis in the family. The deleted allele for marker D6S446 is marked by ‘0’, a recombination event took place between locus D6S1581 and D6S1599 in the mother (II-2).
Genes in the deleted region
The deleted region, spanning approximately 1.2 Mb comprises only four known genes: PDCD2, TBP, PSMB1 and DLL1 and seven putative genes. TCTE3, T-complex-associated testis expressed three gene with male germ-cell-specific expression lies within the breakpoint region. The DLL1 gene, located most proximally to our deletion breakpoint, encodes a human homologue of a Notch delta ligand. The Notch pathway is known to mediate cell to cell communication and plays a role in patterning and differentiation in different tissues.
The PDCD2 gene, programmed cell death 2, contains a zinc-finger MYND domain and is a homologue of the Rp8 gene in rats, where it plays a role in programmed cell death in thymocytes. PSMB1, proteasome subunit, beta type 1, encodes a subunit of the proteasome, a multicatalytic proteinase complex that cleaves peptides in an ATP/ubiquitin-dependent process in nonlysosomal pathways. This gene is tightly linked to the TBP gene in human and in mouse, and is transcribed in the opposite orientation in both species.
TBP encodes the TATA-binding protein, which is a general transcription factor playing a role in the initiation of transcription. TBP contains a long polymorphic CAG repeat in its variable N-terminal domain that corresponds to the polyglutamine region. Expansion of this CAG repeat causes late-onset neurological disorders such as spinocerebellar ataxia 17.15 In addition, the expanded CAG repeat has also been found in patients with Parkinson disease and Huntington disease-like symptoms.16, 17 TBP together with other transcription factors forms a preinitiation complex with polymerase II and binds to the TATA box upstream of the start site of transcription. TBP interacts with TATA-binding protein-associated factors to form the transcription factor IID (TFIID). TFIID binds to the core promoter to position the polymerase properly and serves as the scaffold for assembly of the remainder of the transcription complex.18
Recently, a patient with a deletion smaller than the one in our patients was reported (case 1 by Eash et al.8). This patient lacks only the PSMB1, PDCD2 and TBP genes and presented with features similar to patients with larger 6q subtelomeric deletions.8, 9, 19 Taking all these data together, haploinsufficiency of TBP seemed the most likely cause for the mental retardation present in this family. Several transcription regulators have been linked to neurodevelopmental disorders,20 including genes encoding DNA-binding transcription factors and other transcriptional regulatory proteins such as FMR2, RSK2, MeCP2, ATRX and ZNF41. In most of these genetic defects, it is still unknown how reduced gene expression may lead to neurological dysfunction. In recent years, progress has been made in understanding the molecular mechanisms of the Rubinstein–Taybi syndrome and Rett syndrome caused by mutations in the CBP gene and the MeCP2 gene, respectively. The CBP gene is a transcriptional regulator that binds CREB, a sequence-specific transcriptional activator that relays cAMP signalling, members of the nuclear receptorfamily as well as several other transcriptional regulators. CBP has also histone acetyltransferase activity that makes the chromatin structure more open and thus permissive for transcription of neural plasticity genes. The MeCP2 gene binds and methylates promotors of neural plasticity genes and acts as a transcriptional repressor. Neuronal activity and calcium influx in neural cells activate kinases that phosphorylate MECP2 and relieve expression making the chromatin transcriptionally active.
By using a TBP-specific probe on an MTE assay, we detected high expression of this gene in the cerebral cortex, the frontal, parietal and occipital lobe and the caudate nucleus. Lower expression was observed in the spinal cord, hippocampus, pons, putamen, heart ventricles, testes, lymph node, ileum, ilocaecum and appendix. To verify whether the TBP gene expression was reduced in the patients, we performed real-time PCR with a TBP-specific Taqman® probe on cDNA prepared from lymphocytes of the patients and five controls. In two separate experiments, we detected a relative expression of 0.58±0.12 in the patients (patient III-1: 0.51±0.08; patient III-2: 0.66±0.08, no material of the mother II-2 was available) when compared to controls, indicating the expression of the gene is halved in the patients.
A TBP knockout mouse has been generated previously.21 The murine tbp has been inactivated by homologous recombination after the insertion of a hygromycin resistance cassette into exon 3. tbp−/− mice are not viable and die at the 30–40 cell stage, but heterozygous tbp+/− were fertile and displayed no obvious abnormalities, their weight and size was normal. A group of 11 male tbp+/− mice and 13 control littermates were subjected to a set of behavioural and cognitive assessments. Grip strength and rotarod were similar in both genotypes, indicating that the heterozygotes displayed no obvious motor defects. The open-field test indicated a slight decrease in exploration in the tbp+/− mice, as the entries and time spent in the centre of the open field were reduced (borderline significant with P=0.052 in two-tailed t-test) (Table 2). No differences between the genotypes were observed in entries in the corner of the arena, latencies of first occurrence in the centre of the arena, total path length and maximum distance moved in the arena. In line with these observations, the cage activity test suggested no differences in activity between both genotypes.
Social exploration testing showed the exploratory behaviour was slightly decreased in the group of heterozygotes as the entries in the centre of the open field were reduced (Table 3).
In the elevated plus maze the tbp+/− mice showed an increased number of entries into the open arms (P=0.08) (Table 4), which indicates a tendency towards reduced anxiety.
These results may seem contradictory as the knockout mice have a tendency towards an increased anxiety in the open field test but a tendency towards a decreased anxiety in the elevated plus maze. Contradictory findings in anxiety-related behavioural tests have been reported in other mouse models as well, including in the fmr1 knockout mice.22 It may be that the open-field test, and the elevated plus maze measure different aspects of anxiety.
In the passive avoidance test, seven out of 11 heterozygotes avoided entrance in the dark compartment 24 h after the foot shock, excluding severe damage of working memory. In contrast and surprisingly, 10 of the 13 wild types entered the dark compartment during the 300 s observation period. We do not know what caused this aberrant behaviour in the wild-type littermates, as control mice usually do not have a memory deficit.
Spatial and working memory was assessed using a radial arm maze. In an initial task, all arms were open and baited and the latency to retrieve all food pellets and the number of entries into the arms were counted. Revisits to arms were counted as errors. Both genotypes learned this task equally well and there was no difference in latency or error rate. After a stable response rate, all but one arm were closed to test for working memory. In this task, the mouse was placed on the platform, one arm was open and baited and the mouse learned the position of the baited arm in four trial runs. In a subsequent probe trial, with all arms open but only the previously arm baited, working memory was assessed by measuring latency to enter correct arm and counting the errors of entering unbaited arms. Heterozygote mice displayed an increased latency to retrieve the food bait during the one arm open trials (Figure 3, P=0.053, in ANOVA with repeated measures). However, after 3 days, the latency in both groups becomes similar. This initial delay in latency could indicate a deficit in processing spatial information. (Figure 3a). During the following probe trials, with only the previously baited arm baited but now all arms accessible, wild types and heterozygotes performed equally well in latency and error rate (Figure 3b).

Assessment of spatial working memory in eight-arm radial maze: tbp+/− (open squares) and wild-type (filled circles) mice were trained to retrieve a single food pellet from an open arm of the radial maze. Initially, tbp+/− showed an increased latency to retrieve the food pellet (a), but this difference disappeared over several trial blocks. Values are the means±SEM of four trial runs. In subsequent probe trials, where the same arm was again baited but now all arms were open, there was no difference between the genotypes in latency to retrieve pellet (b) or in number of errors performed (c). *, P≈0.05.
In summary, TBP seems a likely candidate gene for mental retardation in the 6q subtelomeric deletion phenotype. However, we cannot rule out a possible contribution by the other genes in the deleted region, notably PSMB1 and PDCD2 to the phenotype. Therefore, it would be useful to study these genes.
The tbp+/− mice show a trend towards an altered anxiety and working memory deficits. Nonetheless, we have not detected truly significant differences between both genotypes. This may be owing to the small number of mice tested or the genetic background of the mice. As the severity of the mental handicap in patients carrying a 6q subtelomeric deletion varies even within families, for example, the mother in our family was only mildly affected whereas her two sons were severely affected, the phenotype of the knockout mouse may depend upon the genetic background. Therefore, it would be useful to test a larger cohort of tbp+/− mice in a different genetic background to learn more about the possible role of TBP in cognitive development.
References
de Vries BBA, Winter R, Schinzel A, van Ravenswaaij-Arts C : Telomeres: a diagnosis at the end of the chromosomes. J Med Genet 2003; 40: 385–398.
Flint J, Knight S : The use of telomere probes to investigate submicroscopic rearrangements associated with mental retardation. Curr Opin Genet Dev 2003; 13: 310–316.
Rooms L, Reyniers E, Kooy RF : Subtelomeric rearrangements in the mentally retarded: a comparison of detection methods. Hum Mutat 2005; 25: 513–524.
van Bever Y, Rooms L, Laridon A et al: Clinical report of a pure subtelomeric 1qter deletion in a boy with mental retardation and multiple anomalies adds further evidence for a specific phenotype. Am J Med Genet 2005; 135A: 91–95.
Heilstedt HA, Ballif BC, Howard LA et al: Physical map of 1p36, placement of breakpoints in monosomy 1p36, and clinical characterization of the syndrome. Am J Hum Genet 2003; 72: 1200–1212.
Stewart DR, Huang A, Faravelli F et al: Subtelomeric deletions of chromosome 9q: a novel microdeletion syndrome. Am J Med Genet A 2004; 128: 340–351.
Kleefstra T, Smidt M, Banning MJG et al: Disruption of the gene euchromatin histone methyl transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J Med Genet 2005; 42: 299–306.
Eash D, Waggoner D, Chung J, Stevenson D, Martin CL : Calibration of 6q subtelomere deletions to define genotype/phenotype correlations. Clin Genet 2005; 67: 396–403.
Stevenson DA, Brothman AR, Carey JC et al: 6q Subtelomeric deletion: is there a recognizable syndrome? Clin Dysmorphol 2004; 13: 103–106.
Rooms L, Reyniers E, Wuyts W et al: Multiplex ligation-dependent probe amplification to detect subtelomeric rearrangements in routine diagnostics. Clin Genet 2006; 69: 58–64.
Vandesompele J, De Preter K, Pattyn F et al: Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3: 34.1–34.11.
D'Hooge R, Lullmann-Rauch R, Beckers T et al: Neurocognitive and psychotiform behavioral alterations and enhanced hippocampal long-term potentiation in transgenic mice displaying neuropathological features of human alpha-mannosidosis. J Neurosci 2005; 25: 6539–6549.
File SE, Seth P : A review of 25 years of the social interaction test. Eur J Pharmacol 2003; 463: 35–53.
Kraus J, Lederer G, Keri C et al: A familial unbalanced subtelomeric translocation resulting in monosomy 6q27-->qter. J Med Genet 2003; 40: e48.
Koide R, Kobayashi S, Shimohata T et al: A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet 1999; 8: 2047–2053.
Wu YR, Lin HY, Chen CM et al: Genetic testing in spinocerebellar ataxia in Taiwan: expansions of trinucleotide repeats in SCA8 and SCA17 are associated with typical Parkinson's disease. Clin Genet 2004; 65: 209–214.
Stevanin G, Fujigasaki H, Lebre AS et al: Huntington's disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 2003; 126: 1599–1603.
van Roon-Mom WM, Reid SJ, Faull RL, Snell RG : TATA-binding protein in neurodegenerative disease. Neuroscience 2005; 133: 863–872.
Birnbacher R, Chudoba I, Pirc-Danoewinata H et al: Microdissection and reverse painting reveals a microdeletion 6(q26qter) in a de novo r(6) chromosome. Ann Genet 2001; 44: 13–18.
Hong EJ, West AE, Greenberg ME : Transcriptional control of cognitive development. Curr Opin Neurobiol 2005; 15: 21–28.
Martianov I, Viville S, Davidson I : RNA polymerase II transcription in murine cells lacking the TATA binding protein. Science 2002; 298: 1036–1039.
Spencer CM, Alekseyenko O, Serysheva E : Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav 2005; 4: 420–430.
Acknowledgements
Work on the detection of subtelomeric rearrangements in Antwerp was supported by grants from the Marguerite-Marie Delacroix foundation and the Belgian National Fund for Scientific Research–Flanders (FWO). This research was performed in the framework of the Interuniversity Attraction Poles program P5/19 of the Belgian Federal Science Policy Office.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rooms, L., Reyniers, E., Scheers, S. et al. TBP as a candidate gene for mental retardation in patients with subtelomeric 6q deletions. Eur J Hum Genet 14, 1090–1096 (2006). https://doi.org/10.1038/sj.ejhg.5201674
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201674
Keywords
This article is cited by
-
Loss of TAF8 causes TFIID dysfunction and p53-mediated apoptotic neuronal cell death
Cell Death & Differentiation (2022)
-
TAF1, associated with intellectual disability in humans, is essential for embryogenesis and regulates neurodevelopmental processes in zebrafish
Scientific Reports (2019)
-
A child with intellectual disability and dysmorphism due to complex ring chromosome 6: identification of molecular mechanism with review of literature
Italian Journal of Pediatrics (2018)
-
De novo mutations in genes of mediator complex causing syndromic intellectual disability: mediatorpathy or transcriptomopathy?
Pediatric Research (2016)
-
Delineation of candidate genes responsible for structural brain abnormalities in patients with terminal deletions of chromosome 6q27
European Journal of Human Genetics (2015)
