
Abstract
Hypomagnesemia with secondary hypocalcemia (HSH) is a rare inherited disease, characterised by neurological symptoms, such as tetany, muscle spasms and seizures, due to hypocalcemia. It has been suggested that HSH is genetically heterogeneous, but only one causative gene, TRPM6, on chromosome 9 has so far been isolated. We have now studied the genetic background of HSH in four Polish patients belonging to three families, and a HSH patient carrying an apparently balanced X;9 translocation. The translocation patient has long been considered as an example of the X-linked form of HSH. We identified six TRPM6 gene mutations, of which five were novel, in the Polish patients. All the alterations were either nonsense/splicing or missense mutations. The clinical picture of the patients was similar to the HSH patients reported earlier. No genotype–phenotype correlation could be detected. Sequencing did not reveal any TRPM6 or TRPM7 gene mutations in the female HSH patient with an X;9 translocation. Isolation of the translocation breakpoint showed that the chromosome 9 specific breakpoint mapped within satellite III repeat sequence. The X-chromosomal breakpoint was localised to the first intron of the vascular endothelial growth factor gene, VEGFD. No other sequence alterations were observed within the VEGFD gene. Even though the VEGFD gene was interrupted by the X;9 translocation, it seems unlikely that VEGFD is causing the translocation patient's HSH-like phenotype. Furthermore, re-evaluation of patient's clinical symptoms suggests that she did not have a typical HSH.
Similar content being viewed by others

Introduction
Hypomagnesemia with secondary hypocalcemia (HSH; MIM# 602014) is a rare inherited disorder, which leads to affected electrolyte balance during early infancy. The disease is characterised by very low serum magnesium and calcium concentrations. The primary impairment is thought to be in intestinal magnesium absorption (reviewed by Cole and Quamme1), but there is also evidence of defects in renal magnesium handling.2 Patients have neurological symptoms, including tetany, muscle spasms and seizures, due to hypocalcemia. These symptoms can be inhibited by lifelong high oral intakes of magnesium.3 The disease may be fatal if untreated.
It has been suggested that HSH is genetically heterogeneous. The preponderance of reported male patients, and the X;9 translocation female patient with HSH, favoured X-chromosomal recessive inheritance.4, 5 The linkage study by Walder et al6 localised the disease gene to 9q12–q22.2, and recent findings have confirmed that HSH is caused by mutations in an autosomal gene, TRPM6, on 9q21.13.2, 7
The TRPM6 gene consists of 39 exons spanning over a region of 166 kb.2, 7 It is expressed in intestinal epithelia and kidney tubules, with highest expression in distal convoluted tubule (DCT), which is the main site of active transcellular magnesium reabsorption in nephrons. The gene encodes a protein of 2022 amino acids with predicted ion channel domain and carboxy-terminal protein kinase domain. The TRPM6 protein belongs to the transient receptor potential (TRP) channel family,8 with highest sequence similarity to TRPM7. TRPM7 is an ubiquitously expressed calcium and magnesium permeable cation channel.9, 10 TRPM6 and TRPM7 are both unusual bifunctional proteins that contain C-terminal α-kinase domain fused to an ion channel.11 It has been shown that the two proteins interact with each other.12 TRPM6 is unable to form functional channel complexes alone. It requires TRPM7, most probably by the means of hetero-oligomerisation, for trafficking and assembling to the cell surface.
In this study we have analysed the TRPM6 gene in three Polish families with hypomagnesemia and secondary hypocalcemia, as well as isolated the X-chromosomal breakpoint of the female HSH patient with a balanced translocation, t(X;9)(p22;q12).5
Materials and methods
Patients, controls and cell lines
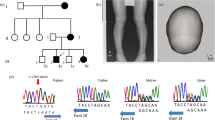
Our study material consisted of four Polish HSH patients, belonging to three families with nonconsanguineous unaffected parents (Figure 1).3 Diagnosis of HSH was based on abnormal low serum calcium and magnesium levels, as well as characteristic neurological symptoms including tremor of extremities followed by seizures and convulsions within 2–6 weeks of birth. The three oldest affected children of the Family 1 had died at young age. DNA was extracted from blood samples using standard procedures. The research followed the tenets of the Declaration of Helsinki.

The pedigrees of three Polish HSH families. Filled squares represent affected males, filled circles affected females, and open symbols unaffected individuals.
A female patient, manifesting HSH and carrying a de novo translocation 46,XX, t(X;9)(p22;q12), was also included in this study.5 In addition to HSH, the patient showed dysmorphic facial features, microcephaly and psychomotor retardation. We used an Epstein–Barr virus-transformed lymphoblastoid cell line, which originated from the female translocation patient. We had also access to DNA, originating from a human–mouse hybrid cell line, which retains the derivative X chromosome as the only human chromosome.13 DNAs from 100 healthy blood donors were used as normal control samples.
Mutation screening
Four Polish HSH patients, and the female HSH patient with a translocation, t(X;9), were included in mutation analysis. All the 39 exons of the TRPM6 gene and seven exons of the VEGFD gene were analysed. The exons and the flanking intronic sequences were PCR-amplified from genomic DNA. Single-stranded conformation analysis (SSCA) was used to detect sequence alterations in amplified exons as described earlier.14 All the exons of the TRPM6 gene were also analysed by direct PCR sequencing (ABI310, Applied Biosystems, Foster City, CA, USA), as well as the exons of the VEGFD gene of the translocation patient. The 39 exons of the TRPM7 gene were sequenced only from the translocation patient.
As not all of the identified sequence alterations showed band shifts in SSCA, in some cases control samples were checked by conformation-sensitive gel electrophoresis15 (mutation c.1437C>A) or mini sequencing16 (mutations c.1060A>C and c.1134+5G>C).
Isolation of the translocation breakpoint
STS mapping
The X-chromosomal translocation breakpoint had previously been mapped between the markers DXS16 and DXS207, DXS43.13 We used the STSs of the YAC contig of this region to narrow down the breakpoint region.17 DNA from the hybrid cell line was used as a PCR template. PCR conditions have been published earlier.17 After the translocation breakpoint had been localised to a one YAC clone, this clone was used as a probe to screen X-chromosome specific cosmid library filters (ICRF).18 New STSs were obtained by sequencing the end-clones of the cosmids of the region. BLAST search was applied to find sequenced clones from Genbank database.
Genome walking
A PCR-based DNA walking method19 was used for detailed identification of the breakpoint (Genome Walker Kit, BD Biosciences, San Jose, CA, USA). Five different adaptor-ligated libraries were constructed of the hybrid cell line DNA according to manufacturer's instructions. Two primers, GSP1 (5′-GTATCTACATCCTCACTCTTCTCTCATTC-3′) and GSP2 (5′-TGGGGAAAGCCAGCCTTCATAAATCTTTT-3′), were designed close to the breakpoint region, which was established by STS mapping. PCR amplifications were performed with GSP1/2 and adaptor primers AP1/2 and the products were sequenced.
FISH analysis
Routine chromosome preparations were made from the lymphoblastoid cell line of the patient. The karyotype of the translocation patient has been published earlier.13 To confirm that the X;9 translocation is the only chromosomal rearrangement, multicolour FISH was performed according to manufacturer's instructions (VYSIS/Abbott Laboratories). FISH studies were also performed to confirm that both a YAC and a cosmid clone, which span over the breakpoint, hybridise to the normal X chromosome and both the translocation X chromosomes. The YAC yWZD2764 and the cosmid ICRFc104O087 were labelled with biotin 11-dUTP by nick-translation (Boehringer Mannheim GmBh, Mannheim, Germany) and hybridisations were performed as described elsewhere.17, 20
Results
Clinical data
Detailed clinical description of the patients is given in Table 1. The clinical picture of the Polish patients did not differ from the HSH patients described earlier.3, 7 However, the clinical picture of the translocation patient was slightly different from HSH. Patient's hypocalcemic convulsions had started only 48 h after birth,5 whereas in HSH patients convulsions appear at the age of two weeks or later.3, 7 Furthermore, in HSH both the erythrocyte and serum Mg2+ concentrations are decreased, while translocation patient's erythrocyte Mg2+ concentration was normal in spite of the decreased serum Mg2+ values.5
Mutation analysis of the TRPM6 and TRPM7 genes
Mutation analysis of the TRPM6 in three Polish families revealed six nucleotide substitutions, of which five were novel (Table 2). All the patients were compound heterozygotes for the TRPM6 gene mutations. Two of the mutations were located within the consensus sequence of the 5′ donor splice site, the other four were in the coding region. None of the six sequence alterations could be seen in two hundred control chromosomes.
The X;9 translocation patient did not have a mutation in the TRPM6 gene. Since TRPM6 can form hetero-oligomeric channel complexes with the TRPM7,12 the TRPM7 gene was also considered as a candidate gene for the disease of the X;9 translocation patient. Sequencing of the coding region of TRPM7 did not, however, reveal mutations.
Breakpoints of the X;9 translocation
Since no TRPM6 or TRPM7 gene mutations were identified in the female HSH patient carrying the balanced t(X;9)(p22;q12) translocation, we decided to isolate the chromosomal breakpoints to see whether the translocation interrupted an important gene. Multicolour FISH analysis confirmed that the metaphases of the lymphoblastoid cell line of the HSH patient contained an X;9 translocation (Ideogram of the translocation is shown in Figure 2). No other structural anomalies could be identified (data not shown).

Partial karyogram showing the X;9 translocation. The location of the TRPM6 gene is shown on chromosome 9.
The X-chromosomal breakpoint had previously been localised between the markers DXS16 and the cluster DXS207, DXS43 on Xp22.13 On chromosome 9, the breakpoint had been mapped between the markers D9S1844 and D9S273, located on 9q12.6 The same human–mouse hybrid cell line was used in this and previous mapping studies. We were able to narrow down the X-chromosomal translocation breakpoint to a small interval with the use of STSs of the YAC contig,17 cosmid clone screening, end clone isolation and BLAST searches. FISH analysis, which was performed on patient's lymphoblastoid cell line, confirmed that both the YAC yWZD2764 and cosmid ICRFc104O087 spanned over the translocation breakpoint in Xp22 (data not shown). The end clones of the cosmid (positive for the marker DXS207) were sequenced to obtain new STSs, which further confirmed that the cosmid spans over the translocation breakpoint. BLAST search revealed another cosmid clone, U27H1 (Genbank accession no. U69570), which was positive for the marker DXS207 and already completely sequenced. Genbank database search showed that the cosmid contained segments of two genes: three last exons and the 3′ utr of PIRIN,21 and the 5′ utr and the first exon of vascular endothelial growth factor gene, VEGFD22, 23, 24 (Figure 3a).

(a) The location of the translocation breakpoint on X chromosome in relation to the YAC clone yWZD2764, the cosmid clone U27H1, the STS markers DXS207 and PIGA-5′, and the genes PIGA, VEGFD and PIR. The picture is not drawn to scale. (b) The translocation breakpoint was localised to the first intron of VEGFD gene. (c) The nucleotide sequences surrounding the translocation breakpoints.
The sequence of the U27H1 cosmid was used to design new PCR primers to further narrow down the translocation breakpoint region. DNA from the hybrid cell line was used as a PCR template. The STSs 6A and 6B were found to be the closest flanking sequences of the breakpoint. The STS 6A could be amplified from the derivative X chromosome and it was located <1 kb from the breakpoint. We continued with Genome walking strategy and identified a PCR product that included both X chromosomal and chromosome 9 specific sequence (Figure 3c). The X-chromosomal translocation breakpoint was localised to the first intron of the VEGFD, 3341 kb distal to the first exon (Figure 3b). The chromosome 9 specific breakpoint mapped within satellite III repeat sequence (5′-AATGG-3′) and thus made it impossible to continue genome walking.
Mutation screening of theVEGFD gene
SSCA screening, followed by direct PCR sequencing, did not reveal any sequence alterations in the coding region of the VEGFD gene of the female HSH patient with an X;9 translocation. The four Polish HSH patients did not have mutations in the VEGFD gene (SSCA screening only).
Discussion
Magnesium homeostasis in mammals depends on the balance between Mg2+ absorption in the intestine and Mg2+ excretion by the kidney. Intestinal magnesium absorption occurs via two different pathways, active transcellular transport and passive paracellular transport.25 The first protein found to be involved in the active magnesium absorption is TRPM6. Immunofluorescence studies have located the TRPM6 protein specifically to the brush-border membrane of the small intestine and the apical membrane of DCT in the kidney,26 which are the main epithelia regulating Mg2+ homeostasis. TPRM6 requires it's closest homolog, TRPM7, for trafficking to the cell membrane.12 It has been shown that TRPM6 interacts directly with TRPM7 to form functional channel complexes on the cell surface. In patients with HSH, mutations in TRPM6 disrupt the transcellular Mg2+ transport. However, treatment with high oral doses of magnesium is sufficient to suppress the symptoms by increasing nonsaturable paracellular magnesium absorption.
In this study, we report five new TRPM6 gene mutations in patients with HSH. Two of the mutations lead to inactivation of the protein, by either destroying a splice site or causing a premature stop codon. The three other mutations were missense mutations leading to amino-acid substitutions. Two of these missense mutations lead to change of amino acid that is conserved between all human TRPM proteins (Figure 4). The transversion c.521T>G causes a hydrophobic isoleucine to be replaced by a hydrophilic arginine. The transition c.2120G>A, causes a sulphur-bond forming cysteine to be replaced by an aromatic tyrosine, and thus probably affects the stability of TRPM6 protein. The third missense mutation, c.1060A>C, results in substitution of a threonine residue, which is conserved only within the closests members of the TRPM family (Figure 4). This mutation potentially leads to a change in bending of the TRPM6 polypeptide in consequence of replacement of uncharged threonine by a cyclic proline. It is still unclear what the definitive effect of these substitutions is on TRPM6 protein. However, all the substitutions were located in the conserved N-terminus of the protein, before the predicted transmembrane domains. It has been shown that the N-terminal TRPM6 mutant S141L disturbs the formation of channel complex with TRPM7 and thus results in loss-of-function of a cation channel.12 Conserved N-terminal T1 domains have been shown to play a critical role in multimerisation and gating of voltage gated K+ channels.27 It could be speculated that the conserved N-terminus in TRPM proteins might have a similar role in channel multimerisation, and therefore amino-acid substitutions in that region could affect the channel complex formation.12

Alignment of the N-terminus of TRPM6 protein with the other human TRPM proteins. Conserved regions are shown by grey (high consensus level, over 90%). Locations of the identified missense mutations are indicated.
Ten of the previously reported twelve TRPM6 mutations, are clearly inactivating splice site or nonsense mutations, which presumably lead to a total loss of function of the encoded channel.2, 7 Only one missense mutation, S141L, has so far been detected,7 and one of the base substitutions (c.3209-68A>G), reported by Walder et al2 as a mutation, turned out to be a single-nucleotide polymorphism (rs2274923) and has recently been corrected in NCBI database. The implications of the new five mutations for the channel function are unclear. We could not detect any genotype–phenotype correlation in our patients. Also, the phenotypes of both our and previously reported patients seemed to be similar.
The translocation patient did not have a mutation in the TRPM6 or TRPM7 gene but we mapped the X;9 translocation breakpoint within the VEGFD gene on X chromosome. VEGFD was isolated based on its homology to other members of the VEGF gene family.22, 23, 24 VEGFs are involved in regulating the formation of blood vessels and lymphatic vessels within the developing embryo and adult. VEGFD protein binds to and induces tyrosine phosphorylation of the endothelial cell receptors VEGFR-2 and VEGFR-3.24 Recent studies have demonstrated that VEGFD can promote angiogenesis, probably due to its ability to activate VEGFR-2, and is capable of inducing lymphangiogenesis, presumably through VEGFR-3.28, 29 VEGFD mRNA is most abundant in the heart, lung, skeletal muscle, colon and small intestine.24
It is now known that TRPM6 gene mutations, which lead to defects in the (re)absorption of magnesium in the intestine and kidney, are causing HSH. Thus, it is difficult to see how VEGFD could be the causative gene for the X;9 translocation patient's hypomagnesemic phenotype. Mutations of VEGFR-3 (the receptor of VEGFD) have been shown to be the underlying cause of congenital hereditary lymphedema.30 The disorder is characterised by chronic swelling of limbs due to dysfunction of lymphatic vessels. The expected phenotype caused by inadequate expression of functional VEGFD would also include dysfunction of lymphatic vessels. Studies of bone formation have clearly indicated that tissue morphogenesis is dependent on the development of, and coordination with, the vascular system.31 Disrupted VEGFD gene might thus have an effect on the dysmorphic features and psychomotor retardation of the translocation patient.
On chromosome 9, the translocation breakpoint is located within satellite III repeat sequences in the constitutive heterochromatin region 9q12. This region contains a mixture of several satellites.32 Satellite III DNA contains a simple 5 bp sequence (5′-AATGG-3′), which is organised into higher order direct repeats of several kb in size. On chromosome 9, satellite III sequences can be found in large quantities in pericentromeric heterochromatin and therefore, the precise translocation breakpoint region could not be determined in this study. Since the translocation breakpoint in our patient is within 5′-AATGG -3′ -repeat, it is unlikely that any gene is interrupted on 9q12 by the translocation. However, the translocation can lead to a position effect, that is, expression of a certain gene near the translocation breakpoint may be altered due to juxtaposition of heterochromatin. Second alternative could be that translocation separates a disease gene from cis-acting regulator elements. Such potential regulatory sequence elements have been identified, for example, as far as to 611 kb upstream and 452 kb downstream of the SOX9 gene.33, 34 Since the TRPM6 gene is located megabases away from the breakpoint,6 it is unlikely that the translocation affects the expression of TRPM6. It must also be noted that we did not identify any sequence alterations in the coding region of the TRPM6 gene of the translocation patient. The recessive nature of the disorder would require that the other copy of the gene should also be affected by mutation. However, in this study whole exon deletions were not excluded. It is possible that an altered expression of some other gene near the breakpoint, due to position effect or separation of cis-acting regulatory elements, could be the cause for the hypomagnesemic phenotype of the translocation patient. There is also a possibility that the phenotype of the translocation patient is caused by a gene independent from translocation.
The X-inactivation status of the patient with X;9 translocation was initially studied using cultured lymphocytes of the patient.5, 35 The derivative X chromosome was found to be late replicating in all cells and the inactivation had not spread to the autosomal 9q segment. Since the normal X chromosome was active, the patient had a functional VEGFD gene in her cells. The selective advantage of the cells, which produce VEGFD growth factor in developing embryo, could be explained by the mitogenic activity, which is induced by VEGFD.24 The intense selective pressure would then be maintaining the severely skewed X-inactivation and keep functional VEGFD active. Since segments of the X chromosome (Xp22-Xpter) in the derivative chromosome 9 cannot undergo X inactivation, this results in functional disomy of distal Xp. The genes residing in this region (which are normally subject to X inactivation) would have an abnormal gene dosage in the cells. Double dosage of Xp22-Xpter genes might contribute to the dysmorphic features and mental retardation of the HSH patient. It must be noted that mental retardation can also result if there is a delay in the diagnosis of HSH and subsequent lack of oral magnesium intake. However, the X;9 translocation patient was already diagnosed at the age of 1 month and properly treated. In the light of the results we obtained from re-evaluating the clinical picture of the translocation patient, it seems very likely that the translocation patient did not have HSH but a disorder similar to HSH.
Accession codes
References
Cole DE, Quamme GA : Inherited disorders of renal magnesium handling. J Am Soc Nephrol 2000; 11: 1937–1947.
Walder RY, Landau D, Meyer P et al: Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet 2002; 31: 171–174.
Pronicka E, Gruszczynska B : Familial hypomagnesemia with secondary hypocalcaemia – autosomal or X-linked inheritance. J Inher Metab Dis 1991; 14: 397–399.
Teebi AS : Primary hypomagnesaemia, an X-borne allele? Lancet 1983; 1: 701.
Meyer M, Mattei JF, Viallard JL, Goumy P, Dastugue B, Malpuech G : Hypocalcemie magnesodependante par trouble specifique de l’absorption du magnesium, associee a une anomalie chromosomique. Rev Franc Endocrinol Clin 1978; 19: 101–108.
Walder RY, Shalev H, Brennan TMH et al: Familial hypomagnesemia maps to chromosome 9q, not to the X chromosome: genetic linkage mapping and analysis of a balanced translocation breakpoint. Hum Mol Genet 1997; 6: 1491–1497.
Schlingmann KP, Weber S, Peters M et al: Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet 2002; 31: 166–170.
Montell C, Birnbaumer L, Flockerzi V : The TRP channels, a remarkably functional family. Cell 2002; 108: 595–598.
Runnels LW, Yue L, Clapham DE : TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 2001; 291: 1043–1047.
Nadler MJ, Hermosura MC, Inabe K et al: LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 2001; 411: 590–595.
Riazanova LV, Pavur KS, Petrov AN, Dorovkov MV, Riazanov AG : Novel type of signaling molecules: protein kinases covalently linked to ion channels. Mol Biol (Mosk) 2001; 35: 321–332.
Chubanov V, Waldegger S, Mederos Y et al: Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci USA 2004; 101: 2894–2899.
Chery M, Biancalana V, Philippe C, Malpuech G, Hilgenkrantz S, Hanauer A : Hypomagnesemia with secondary hypocalcemia in a female with balanced X;9 translocation: mapping of the Xp22 chromosome breakpoint. Hum Genet 1994; 93: 587–591.
Huopaniemi L, Rantala A, Forsius H, Somer M, de la Chapelle A, Alitalo T : Three widespread founder mutations contribute to high incidence of X-linked juvenile retinoschisis in Finland. Eur J Hum Genet 1999; 7: 368–376.
Ganguly A, Rock MJ, Prockop DJ : Conformation-sensitive gel electrophoresis for rapid detection of single-base differences in double-stranded PCR products and DNA fragments: evidence for solvent-induced bends in DNA heteroduplexes. Proc Natl Acad Sci USA 1993; 90: 10325–10329.
Syvänen AC, Sajantila A, Lukka M : Identification of individuals by analysis of biallelic DNA markers, using PCR and solid-phase minisequencing. Am J Hum Genet 1993; 52: 46–59.
Alitalo T, Francis F, Kere J, Lehrach H, Schlessinger D, Willard HF : A 6-Mb YAC contig in Xp22.1-p22.2 spanning the DXS69E, XE59, GLRA2, PIGA, GRPR, CALB3, and PHKA2 genes. Genomics 1995; 25: 691–700.
Lehrach H, Drmanac J, Hoheisel J et al: Hybridization fingerprinting in genome mapping and sequencing. In Davies KE, Tilghman S (eds): Genome analysis: genetic and physical mapping. New York: Cold Spring Harbor, 1990, pp 39–81.
Siebert PD, Chenchik A, Kellogg DE, Lukyanov KA, Lukyanov SA : An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res 1995; 23: 1087–1088.
Pinkel D, Straume T, Gray JW : Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci USA 1986; 83: 2934–2938.
Wendler WM, Kremmer E, Forster R, Winnacker EL : Identification of pirin, a novel highly conserved nuclear protein. J Biol Chem 1997; 272: 8482–8489.
Yamada Y, Nezu J, Shimane M, Hirata Y : Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics 1997; 42: 483–488.
Rocchigiani M, Lestingi M, Luddi A et al: Human FIGF: cloning, gene structure, and mapping to chromosome Xp22.1 between the PIGA and the GRPR genes. Genomics 1998; 47: 207–216.
Achen MG, Jeltsch M, Kukk E et al: Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA 1998; 95: 548–553.
Kerstan D, Quamme G : Physiology and pathophysiology of intestinal absorption of magnesium. In Massry SG, Morii H, Nishizawa Y (eds): Calcium in Internal Medicine. London: Springer-Verlag, 2002, pp 171–183.
Voets T, Nilius B, Hoefs S et al: TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem 2004; 279: 19–25.
Minor DL, Lin YF, Mobley BC et al: The polar T1 interface is linked to conformational changes that open the voltage-gated potassium channel. Cell 2000; 102: 657–670.
Rissanen TT, Markkanen JE, Gruchala M et al: VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res 2003; 92: 1098–1106.
Stacker SA, Caesar C, Baldwin ME et al: VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med 2001; 7: 186–191.
Karkkainen MJ, Ferrell RE, Lawrence EC et al: Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet 2000; 25: 153–159.
Vu TH, Shipley JM, Bergers G et al: MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 1998; 93: 411–422.
Gosden JR, Mitchell AR, Buckland RA, Clayton RP, Evans HJ : The location of four human satellite DNAs on human chromosomes. Birth Defects Orig Artic Ser 1975; 11: 168–169.
Bagheri-Fam S, Ferraz C, Demaille J, Scherer G, Pfeifer D : Comparative genomics of the SOX9 region in human and Fugu rubripes: conservation of short regulatory sequence elements within large intergenic regions. Genomics 2001; 78: 73–82.
Pop R, Conz C, Lindenberg KS et al: Screening of the 1 Mb SOX9 5′ control region by array CGH identifies a large deletion in a case of campomelic dysplasia with XY sex reversal. J Med Genet 2004; 41: e47.
Mattei MG, Mattei JF, Ayme S, Malpuech G, Giraud F : A dynamic study in two new cases of X chromosome translocations. Hum Genet 1978; 41: 251–257.
Acknowledgements
Part of this study was performed at the Folkhälsan Institute of Genetics. This study was supported by the Helsinki University Hospital Grants TYH8301 and TYH1338, the European Communities Grant PL950889, Sigrid Juselius Foundation and Jenny and Antti Wihuri Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jalkanen, R., Pronicka, E., Tyynismaa, H. et al. Genetic background of HSH in three Polish families and a patient with an X;9 translocation. Eur J Hum Genet 14, 55–62 (2006). https://doi.org/10.1038/sj.ejhg.5201515
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201515
Keywords
This article is cited by
-
Intestinal hypomagnesemia in an Iranian patient with a novel TRPM6 variant: a case report and review of the literature
CEN Case Reports (2023)
-
New TRPM6 missense mutations linked to hypomagnesemia with secondary hypocalcemia
European Journal of Human Genetics (2014)
