
Abstract
Wolfram syndrome (WS) is a neuro-degenerative autosomal recessive (AR) disorder (OMIM #222300) caused by mutations in the WFS1 gene on 4p16.1. More than 120 mutations have been identified in WFS1 associated with AR WS, as well as autosomal dominant nonsyndromic low-frequency sensorineural hearing loss (LFSNHL). WFS1 variants were identified in eight subjects from seven families with WS, leading to the identification of four novel mutations, Q194X (nonsense), H313Y (missense), L313fsX360 (duplication frame shift) and F883fsX951 (deletion frame shift), and four previously reported mutations, A133T and L543R (missense), V415del (in frame triple deletion) and F883fsX950 (deletion frame shift). A mutation was found in 11/14 disease chromosomes, two subjects were homozygous for one mutation, one subject was compound heterozygous for two nucleotide substitutions (missense), one subject was compound heterozygous for a duplication and a deletion (frame shift), and in three families only one mutation was detected (Q194X and H313Y). All affected individuals shared clinically early-onset diabetes mellitus and progressive optic atrophy with onset in the first and second decades, respectively. In contrast, diabetes insipidus was present in two subjects only. Various degrees and types of hearing impairment were diagnosed in six individuals and cataract was observed in five subjects.
Similar content being viewed by others

Introduction
Wolfram syndrome (WS) was first described by Wolfram and Wagener in 1938 as the association of childhood-onset diabetes and optic atrophy (OA).1 The acronym, DIDMOAD, is often used synonymously to WS in order to include four characteristic clinical abnormalities: diabetes insipidus, diabetes mellitus (DM), OA, and deafness, among which childhood-onset DM and OA often are considered sufficient for a diagnosis of WS. Since the original delineation the clinical spectrum of WS has, however, been recognized to comprise a multi-system neuro-degenerative disorder, which in addition to the classical four components includes ataxia, behavioural and psychiatric illness, apnoea, hypogonadism, and renal tract atonia with hydronephrosis. Mutations have been reported in a gene, WFS1, which give rise to two distinct phenotypes: autosomal recessive (AR) WS and the autosomal dominant low-frequency sensorineural hearing loss (AD LFSNHL).2, 3, 4 Considerable clinical pleiomorphism, the use of different diagnostic criteria, and a different focus on clinical details among published reports make it difficult to compare the frequency of associated abnormalities. Furthermore, a second genetic locus on chromosome 4q has been reported accounting for additional clinical variation including peptic ulcer and a platelet aggregation defect.5, 6 However, the growing number of WS reports with confirmed WFS1 mutations and the observation of a correlation between missense WFS1 mutations and variations in phenotypes offer a basis for establishing a better understanding of genotype–phenotype correlation.7, 8
The WFS1 gene, which was mapped in 19949 and cloned in 1998,2, 10 encodes an 890 amino-acid (aa) endoplasmatic reticulum-membrane-located protein, wolframin. It has nine predicted α-helical transmembrane segments and otherwise no homology to any other recognized protein domains.2 No ‘classical’ sorting signals can be identified and it is composed of a cytoplasm located N-terminal and an ER lumen located C-terminal, but the molecular function has not been established.11, 12, 13, 14 The majority of the WFS1 mutations are located either in the transmembrane domains or in the C-terminal of the protein, and the nature of the mutations seems to have clinical implications. Only missense mutations are found together with AD LFSNHL, but both missense, nonsense and frame shift mutations are present in AR WS and infer that WS is the result of a complete lack of functional wolframin protein.8 Functional studies of the WFS1 protein have been published, but the mechanism of its biological function and the pathophysiological basis is not understood. Expression of wild-type or mutated WFS1 protein in Xenopus oocytes induced a novel Ca2+ ion channel-type activity over the ER membrane,12 and other studies demonstrated both the orientation in the membrane, N-glycosylation of the C-terminal part and an oligomerization of the WFS1 protein, most likely as a homotetramer, in the ER membrane.13, 14 In the present study, molecular analysis of the WFS1 gene in seven families with WS resulted in identification of eight different mutations; one of these was a de novo mutation occurring independently in two families, whereas the remaining ones were inherited.
Materials and methods
Patients and families
Patient records were retrieved from the files of The National Eye Clinic for the Visually Impaired (NEC – http://www.visaid.dk/) among referrals from the last 25 years. The minimal diagnostic criteria for WS were early-onset DM and progressive OA with insidious onset during the first or second decade. All subjects underwent an ophthalmological examination including slit lamp assessment and ophthalmoscopy with dilated pupils. Additional examinations included dynamic visual field measurements with the Goldmann perimeter, colour vision tests, and visual evoked potentials. Full-field electroretinography (ERG) was performed in one patient (EE32:II.3). The patients were followed at NEC for various periods of time between two and 24 years. Additional medical information was obtained from records at the departments of ophthalmology, paediatrics, and audiology. Pure tone audiograms were undertaken only in patients who had symptoms of hearing impairment. The files contained only scanty information about family members and this study did not include a systematic examination of all family members with regard to vision, endocrine, and hearing function. No effort was made to retrieve data on psychiatric disorders or minor manifestations of WS.
Venous blood was drawn from all patients as well as their parents and siblings if available. The study adhered to the tenets of the declaration of Helsinki and all subjects gave written consent to participate in the study.
DNA extraction
Genomic DNA was extracted from whole blood using standard procedures. Screening of mutations and polymorphisms in the normal population was carried on 60 independent persons selected from the Copenhagen Family Bank.15
PCR amplification and DNA sequencing of the WFS1 gene
All eight exons, the exon–intron sequences, and the minimal promoter were sequenced in one affected member of five families; EE31:II.2, EE32:II.3, EE33:II.2, EE76:II.2, EE77:II.1, and in two affected individuals in one family, EE34:II.2 and EE34:II.4. The family members EE53:I.1 and EE53:II.2 were sequenced in the 5′ part of exon 8 (the family has previously been analysed, unpublished data not presented). Both strands were sequenced and aligned to the reference sequence GenBank accession no. AF084481 and AC116517. A minimum of 100 base pairs (bp) of exon–intron boundaries and at least 120 bp of the minimal upstream promoter region and 350 bp of the noncoding 3′UTR were analysed. Mutations or single-nucleotide polymorphisms (SNPs) and zygotic status for each change were noted and mutations confirmed by restriction enzyme digests. A total of 22 primers were used (Table 1) and PCR was carried out under standard conditions according to the enzyme manufacturers' protocols; Taq DNA polymerase was purchased from Amersham (Taq DNA Polymerase), New England Biolabs (USA); AmpliTaq Gold DNA Polymerase from Applied Biosystem Industries (USA); and Platinum Taq DNA polymerase from Invitrogen (USA). Reactions were carried out in 15 μl volumes containing buffer according to the manufacturers' protocols: 2.5 μ M dNTP, 10 μ M of each primer, 0.008% cresol red (Aldrich), 12% sucrose (w/v), and 50–100 ng template DNA. Standard reaction conditions for all primers were 95°C for 5 min; then 40 cycles at 95°C for 30 s, 56.4°C for 30 s, and 72°C for 1 min, followed by 5 min at 72°C. PCR reactions were analysed by 2% agarose gel electrophoresis by staining with ethidium bromide, 1 × TBE, before sequencing. Both strands were sequenced directly using the PCR primers and BigDye version 1.0 sequencing kit (Applied Biosystems) without further modifications on an ABI377 sequenator. Sequence data were analysed using standard software and sequence alignments were carried out using ClustalW or BLAST, respectively.16, 17
Control digests of identified mutations and screening of control population
Each identified mutation was confirmed by restriction enzyme digest (New England Biolabs, USA); standard restriction enzyme digests were carried out using 3–5 μl PCR products in 20 μl volumes. A mismatch primer, 5′-TCAACCCCAAGAAGAAGACG, was constructed for detection of the mutation c.580C>T in family EE31 and digested with HgaI (PCR product generated by the mismatch primer and WFS1-ex5-R), the mutation c.397G>A in family EE32 was detected after digest with the endonuclease Fnu4HI (primer pair WFS1-ex4-F and WFS1-ex4-R), the mutation c.1628T>G also found in family EE32 was detected by cleavage with the endonuclease SacI (primer pair WFS1_8-2F and WFS1_8-2R), the mutation c.937C>T in family EE33 was detected by digest with the endonuclease RsaI (primer pair WFS1_8-1F and WFS1_8-1R) and in family EE77 with HpyCH4V (same primer pair). Restriction enzyme digests with MboI confirmed the mutation c.2649delC in family EE34 (primer pair WFS1_8-4F and WFS1_8-4R), the mutation c.937–941dupCACTG in family EE76 was detected by digest with the endonuclease BsgI (primer pair WFS1_8-1F and WFS1_8-1R), and the mutation c.2646–2649delTTTC was detected by digest with the enzyme MboII (primer pair WFS1_8-4F and WFS1_8-4R). Digest of 60 normal control persons was carried out for all new mutations.
Paternity test for family EE33 and EE77
Standard conditions for 33P-end-labelling of the forward PCR primers were used; gamma-33P-ATP was purchased from Hartmann Analytic (Germany) and T4-DNA-polynucleotide kinase from Fermentas (Lithuania). PCR standard conditions using the end-labelled PCR primer without further purification were carried out under the following conditions: 95°C, 20 s, 55°C, 20 s, and 72°C for 30 s for 35 cycles using New England Taq-DNA polymerase (USA). For family EE33 STS markers D11S922, D13S170, D13S154, D2S925, D12S78, D12S1720, D13S328 and for family EE77, marker systems D14S273, D14S63, D14S611, D7S661, D7S798, D7S2447, D7S2207, D7S1824, and D10S1697 were used.
Results
Patients
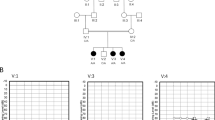
Ten patients, seven females and three males from seven families (Figure 1) fulfilled the minimal diagnostic criteria. Consanguinity was known in one family (EE53), the parents being first-degree cousins. The main clinical data are summarized in Table 2, from which it appears that DM was diagnosed in the first decade between 14 months and 8 years of age, followed by the recognition of OA between 1 and 15 years later, a delay of 5.3 years in average. The OA had an insidious onset and may have been present for some time until the visual acuity (VA) was affected. After the diagnosis of DM, most patients had yearly eye examinations and initially a normal VA was measured. OA was not suspected and pallor of the optic discs was not noticed before the corrected VA had dropped to 0.5. The OA had a slowly progressive course and resulted in a very low VA <0.02 in patients with follow-up periods of 20–25 years. Diabetic retinopathy was diagnosed in EE31:II.2 after 22 years of diabetes. Despite argon laser photocoagulation, she developed a proliferative retinopathy with vitreous hemorrhages during the following years. Another patient, EE34:II.2, did not develop retinopathy during 29 years of observation following the diagnosis of DM. Diabetes insipidus was diagnosed in two patients of ages 8 and 11 years, and various degrees and types of hearing impairment were noticed in six patients (Table 2). The remaining four subjects had no hearing difficulties and audiological examination was not performed. Among other manifestations, a high prevalence of cataract was striking. All cataracts were bilateral, but otherwise they varied with respect to type and degree. Cataracts were observed in five individuals from families EE31, EE33, EE34, EE76, and EE77, respectively. The lenticular opacities had mainly a posterior subcapsular location, but scattered cortical elements were also noted. The cataracts were visually insignificant in three subjects, whereas two of the probands underwent extracapsular cataract extraction on both eyes. One of these, EE76:II.2, developed cataracts subacutely at the age of 9 years during a period of 3 months. Refractive errors varied between – 4.0 Diopters (D) and +1.5 D, with one exception: patient EE31:II.2 had refractive values of +7.75–1.25 × 180° and +8.75–1.75 × 15° in her right and left eye, respectively. Apart from attenuated optic nerves, MRI was normal in two subjects, EE53:II.2 and EE76:II.2, and revealed a slender medulla oblongata in patient EE34:II.2. Two patients, EE53:II.1 and EE77:II.1, were developmentally retarded. The cause of death was unknown in two of the three patients who died during the observation period. An autopsy on patient EE53:II.1 who died from pulmonary insufficiency due to fulminant pneumonia showed OA with severe loss of myelinated nerve fibres, distorted axons with bulbs or torpedo-like formations, and astrogliosis. Hypothalamus had axonal bulbs and torpedos with slight microgliosis, and the spinal cord showed axonal bulbs and amyloid precursor protein (APP) accumulation in the pyramidal tract. The brain stem and pons were smaller than average, but no definitive olivo-ponto-cerebellar atrophy (multiple system atrophy), as has been described in WS (Henning Laursen, personal communication).

Pedigrees of seven Danish WS families. (a) Pedigrees for the WS families; a denotes completely sequenced patients; b denotes patients where only the mutated region is sequenced; c denotes patients analysed by restriction enzyme digest; (b) Digested PCR products separated by gel electrophoresis; family EE31: digest with HgaI, normal allele 260 bp, c.580C>T allele 234 bp; family EE32: c.397G>A digested with Fnu4HI, normal allele 73 bp, mutated allele 61 bp, and c.1628T>G digested with SacI, normal allele 416 bp, mutated allele 569 bp; family EE33: digest with RsaI, normal allele 542 bp, mutant allele 509 bp; family EE34: digest with MboI, normal allele 450 bp, mutated allele 495 bp; family EE76: c.937–941dupCACTG digested with BsgI, normal allele 151 and 200 bp, mutant allele additional 62 and 143 bp, and c.2646–2649delTTTC digested with MboII, normal allele 200 bp and mutant allele 185 bp; family EE77: digest with HpyCH4V, normal allele 68 and 143 bp, mutated allele 200 bp. Size marker M: 50 or 100 bp DNA ladder.
Family and WFS1 mutations analysis
Family EE31
One novel nonsense mutation, c.580C>T, Q194X, was found in exon 5. The nucleotide substitution converts the glutamine CAG codon into the stop-codon TAG (Table 3). The remaining 193 aa of the N-terminal represent a truncated WFS1 protein (Figure 2a), which is likely to be degraded after translation. DNA was available only from one family member, EE31:II.2 (Figure 1a), and no second mutation was identified in the WFS1 coding regions. Micro-deletions of all exons except exon 7 could be excluded based on the pattern of polymorphisms (Table 4).

Mutations and polymorphisms in relation to the primary and secondary structure of wfs1. (a) The cytoplasmatic regions are yellow, the endoplasmatic regions are green and the transmembrane α-helices are blue, the mutations are red and nonsynonymous amino-acid changes are blue. (b) A total of 15 mutation-surrounding amino acids from six different species. The transmembrane domains are boxed; identical amino acids are marked by an asterisk, and conserved amino acids by a colon. Human – Homo sapiens, NP_005996; chimp – Pan troglodytes, protein prediction from contig AADA1090020; rat - Rattus norvegicus, NP_114011; mouse – Mus musculus, NP_035846; chick – Gallus gallus, protein prediction from contig AADNO1046637; and fugu – Takifugu rubripes, protein prediction from scaffold_2015.35, 36
Family EE32
Two missense mutations were identified in patient EE32:II.3. The mutation c.397G>A, A133T, located in exon 4, is known to cause WS18, 19 and change a small nonpolar alanine (GCC) in the central part of the cytoplasmatic domain into a hydrophilic polar uncharged threonine (ACC, Figure 2a). The second mutation c.1628T>G, L543R, is located in exon 8 and changes a hydrophobic nonpolar leucine (CTC) in the sixth transmembrane domain into a polar charged basic arginine residue (CGC, Figure 2a). L543R is known to cause WS20 most likely due to impairment of a proper insertion of the protein into the ER membrane. Analysis in the parents disclosed A133T to be paternal and L543R to be maternal (Figure 1a, data not shown).
Family EE33
Only one mutation, c.937C>T, H313Y, located in exon 8 was identified in person EE33:II.2. The mutation is novel and changes the basic polar charged histidine (CAC) located in the border region of the first transmembrane domain into the aromatic bulky polar uncharged tyrosine (TAC). This amino-acid change may impair integration of WFS1 protein into the ER membrane (Figure 2a). The mutation was not found in any of the parents, for which reason H313Y may be considered a de novo mutation (Figure 1b). Proof of paternity was established by seven different STS marker systems (see materials and methods). Micro-deletions of all exons except exon 7 can be excluded based on polymorphism pattern.
Family EE34
The novel mutation c.2649delC, F883fsX953, was found on both alleles in persons EE34:II.2 and EE34:II.4. The mutation leads to a frame shift and a new 951 aa WFS1 protein product, that is expanded in the ER-located domain. The mutation may interfere with transport across and integration into the ER membrane. The frame shift c.2649delC mutation is located in the polythymine repeat 19 nucleotides from the genuine stop codon and the consequence of the mutation is analogous to c.2646–2649delTTTC (F883fsX950) found in EE76 and other mutations reported in the polyphenylalanine repeat causing WS.10, 18, 19 Analysis of the family revealed that one sister (EE34:II.1) and both parents (EE34:I.1 and EE34:I.2) were heterozygous carriers, whereas one sister (EE34:II.6) harboured the wild-type allele on both chromosomes (Figure 1b).
The two patients, EE34:II.2 and EE34:II.4, showed no DNA differences and might be related, despite our missing family information about a possible parental consanguinity (Tables 2 and 4).
Family EE53
Patient EE53:I.1 was heterozygous and EE53:II.2 was homozygous for the mutation c.1243–1245delGTC, V415del, in accordance with the known parental consanguinity (Figure 1b). We have deduced the carrier state of the deceased mother who was unavailable for mutation analysis (Figure 1). The mutation is located in exon 8 and the deleted amino acid positioned in the third transmembrane domain. V415del is previously described in combination with WS,20, 21 and most likely affects integration of wfs1 into the membrane.
Family EE76
EE76:II.2 was compound heterozygous, carrying the mutations c.937–941dupCACTG, L313fsX360 and c.2646–2649delTTTC, F883fsX950 in trans (Figure 1b, Table 3). The c.937–941dupCACTG mutation was found in the father and brother, both healthy, whereas c.2646–2649delTTTC mutation was only found in the affected person. The deceased mother was unavailable for mutation analysis. The 5-bp duplication results in a frame shift at L315 and extension of wfs1 into a 359 aa fusion protein, which presumably is degraded after translation. The four base deletion results in a frame shift at F883 and a 949 aa fusion protein, and the mutation is analogous to the c.2649delC mutations in family EE34 and other reported WS mutations in the polyphenylalanine.10, 18, 21
Family EE77
EE77:II.1 carried the mutation, c.937C>T, H313Y, which is identical to the mutation identified in the unrelated family EE33. None of the parents carried this mutation (Figure 1b) implicating a de novo mutation, and no other mutation was detected in the WFS1 coding regions. Proof of paternity was established by 12 different STS marker systems (see materials and methods).
Single-nucleotide WFS1 polymorphisms
Small deletions or SNPs were identified both in intronic and exonic regions in five of the seven families (Table 4). In family EE34, the two sequenced patients were homozygous and in the consanguineous family EE53 the affected patient was homozygous in the sequenced regions. In total, 12 SNPs were found in all introns except intron 5, of which three have not previously been described (Table 4). No polymorphisms were found in the minimal promoter or in the 5′UTR region and three known polymorphisms were found in the 3′UTR. Seven known SNPs, one in exon 6 and six in exon 8, were identified in the coding sequence; five do not result in amino-acid changes and two result in amino-acid changes (Table 4).
Discussion
We have undertaken mutation analysis in seven Danish families with WS and identified a mutation in 11/14 disease chromosomes (78.6%), which is comparable with other reports.8 Two families were compound heterozygous (EE32 and EE76), two families were homozygous (EE34 and EE53), and in three families it was only possible to identify one mutation (EE31, EE33, and EE77).
Four mutations, Q194X, L313fsX360, F883fsX951, and H313Y, have not been reported before. The first 3 mutations are likely to result in an impaired protein function, whereas the functional consequences of the missense mutation are uncertain. The mutation H313Y in the TM1 domain (Figure 2a) has not been reported before, but other mutations in TM1 (M312R and A326V) have been reported in subjects with psychiatric disorder.22, 23
Figure 2b shows alignment of WFS1 from six different organisms, two primates, two rodents, one bird, and one fish. The overall conservation for WFS1 between the species is 99–56%, and higher in the ER lumen C-terminal than in the cytoplasmatic N-terminal (data not shown). All the identified mutations affect conserved amino acids among the six organisms in the WFS1, protein with the exception of two that differs in Fugu (Figure 2b).
Identification of a mutation in the WFS1 coding region in only one allele in three of the families implies that a second disease-causing mutation has escaped detection (Table 2).
The missing mutation could be one of the identified polymorphisms leading to either decreased stability or translation of the mRNA product or alternative splice forms of the WFS1 mRNA, both phenomena leading to either a nonfunctional or an absent WFS1 protein. The polymorphisms V395V, H611R, K811K, and S855S found in EE31 and EE33 are all associated with psychiatric disorders alone or in combination with either DM and sensorineural hearing impairment, DM type I, or DM type II.24, 25, 26, 27 The cis- or trans-location of the identified polymorphisms as well as their possible influence on phenotypes are unknown. The SNP Ivs4−15T>C is located so close to the intron4–exon5 border, that it might interfere with the intron–exon splicing. This SNP is heterozygous in family EE31, EE33, and EE77, but also in the compound heterozygous family EE76. A second mutation may also be located in the promoter regions or in other cis- or trans-acting regulatory elements. Furthermore, mutations may also occur in the second WS locus, WFS2, at 4q22–q24.6
The occurrence of the mutation H313Y de novo in two unrelated families (EE33 and EE77) is a surprising finding, considering the fact that de novo mutations seem to be very rare in WSF1.8 The phylogenetic conservation of H313 (Figure 2b) and the restriction enzyme RsaI analysis of 60 control persons support the causative role of the sequence change, c.937C>T. Hypothetically, H313Y may act as a dominant-negative mutation analogous to dominant mutations in LFSNHL. WS mutations are in general inactivating ‘loss-of-function’ mutations as nonsense or frame shift mutations,8 whereas mutations found in combination with AD LFSNHL are small noninactivating ‘gain-of-function’ mutations.28
In spite of selection criteria precluding cases of WS without DM or OA, considerable phenotypic variability is noticed even among the few cases in the present material. However, we found less intrafamilial variation than between patients from different families.
In addition to the classical quartet defining the DIDMOAD syndrome, our sample included patients with neuro-endocrine abnormalities, including growth retardation, delayed puberty, and primary amenorrhoea. The possibility of a normal reproductive function was illustrated by the fact that one female and two males over the age of 25 years had children. The presence of cataract in five patients is in line with a growing number of reports indicating that cataract should be considered a more regular part of WS than hitherto recognized.29, 30, 31, 32
No firm correlation between genotypes and phenotypes has emerged so far despite the large number of reported WS subjects with specified WFS1 mutations. Hardy et al21 reported on four British families with a 4-bp deletion at position 2648 (2646 in the nomenclature used by us) and suggested that brain stem atrophy with severe respiratory involvement was associated with this homozygous mutation, resulting in a chimeric 949 aa wfs1 protein.21, 33 In fact, a cerebral MRI in EE34:II.3 with a similar homozygous 1-bp mutation showed a slender brain stem, but no respiratory symptoms were reported in any of the two affected brothers in this family. In addition to severe OA, they had well-regulated DM without signs of diabetic retinopathy after 37 and 28 years duration, but neither hearing impairment nor diabetes insipidus or other neuro-endocrine abnormalities. Since both affected persons in family EE34 thus demonstrated a mild WS phenotype, it is tempting to hypothesize that the extended chimeric wfs1 found in EE34 may retain part of its function, resulting in milder symptoms in the homozygous state.
None of the present probands had the mt-DNA A3243G mutation or the A1555G mutation (data not shown), which has been postulated to possibly influence the severity of the individual WFS1 mutations in different patients. Our negative findings are in line with the absence of mtDNA mutations reported by others.34, 35
Audiometry on the WS patient from family EE32 with two missense mutations in exon 4 and exon 8 showed HL of 30 dB at 6000 Hz and 60 dB at 8000 Hz. This subject was one of two with DI. The second DI patient, EE76:II.2, also had hearing impairment affecting primarily the high frequencies (HF) hearing deficiency. Both affected sisters in family EE53 had hearing impairment affecting low and high frequencies, but better preservation of hearing in the mid-frequencies. Their hearing problems, however, might in part be conductive due to middle ear pathology. Both probands with the de novo H313Y mutation, EE33 and EE77, had congenital profound hearing impairment, but only one of them, EE77:II.1, had mental retardation, again showing clinical variability. It would be of great prognostic significance to be able to assess the functional implications of different WFS1 mutations along with increasing understanding of the normal function of the wolframin protein.
References
Wolfram DJ, Wagener HP : Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Mayo Clin Proc 1938; 13: 715–718.
Strøm TM, Hortnagel K, Hofmann S et al: Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet 1998; 7: 2021–2028.
Bespalova IN, Van Camp GJH, Bom S et al: Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low-frequency sensorineural hearing loss. Hum Mol Genet 2001; 10: 2501–2508.
Young TL, Ives E, Lynch E et al: Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 2001; 10: 2509–2514.
Ajlouni K, Jarrah N, El-Khateeb M, El-Zaheri M, El Shanti H, Lidral A : Wolfram syndrome: identification of a phenotypic and genotypic variant from Jordan. Am J Med Genet 2002; 115: 61–65.
El-Shanti H, Lidral AC, Jarrah N, Druhan L, Ajlouni K : Homozygosity mapping identifies an additional locus for Wolfram syndrome on chromosome 4q. Am J Hum Genet 2000; 66: 1229–1236.
Gómez-Zaera M, Strom TM, Rodriguez B, Estivill X, Meitinger T, Nunes V : Presence of a major WFS1 mutation in Spanish Wolfram syndrome pedigrees. Mol Genet Metab 2001; 72: 72–81.
Cryns K, Sivakumaran TA, Van den Ouweland JM et al: Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum Mutat 2003; 22: 275–287.
Polymeropoulos MH, Swift RG, Swift M : Linkage of the gene for Wolfram syndrome to markers on the short arm of chromosome 4. Nat Genet 1994; 8: 95–97.
Inoue H, Tanizawa Y, Wasson J et al: A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 1998; 20: 143–148.
Takeda K, Inoue H, Tanizawa Y et al: WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 2001; 10: 477–484.
Osman AA, Saito M, Makepeace C, Permutt MA, Schlesinger P, Mueckler M : Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J Biol Chem 2003; 278: 52755–52762.
Hofmann S, Philbrook C, Gerbitz KD, Bauer MF : Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet 2003; 12: 2003–2012.
Takeda K, Inoue H, Tanizawa Y et al: WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 2001; 10: 477–484.
Eiberg H, Nielsen LS, Klausen J et al: Linkage between serum cholinesterase 2 (CHE2) and (-crystalline gene cluster (CRYG): assignment to chromosome 2. Clin Genet 1989; 35: 313–321.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ : Basic local alignment search tool. J Mol Biol 1990; 215: 403–410.
Thompson JD, Higgins DG, Gibson TJ : CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673–4680.
Khanim F, Kirk J, Latif F, Barrett TG : WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum Mutat 2001; 17: 357–367.
Giuliano F, Bannwarth S, Monnot S et al: French Group of WS. Wolfram syndrome in French population: characterization of novel mutations and polymorphisms in the WFS1 gene. Hum Mutat 2005; 25: 99–100.
Colosimo A, Guida V, Rigoli L et al: Molecular detection of novel WFS1 mutations in patients with Wolfram syndrome by a DHPLC-based assay. Hum Mutat 2003; 21: 622–629.
Hardy C, Khanim F, Torres R et al: Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am J Hum Genet 1999; 65: 1279–1290.
Torres R, Leroy E, Hu X et al: Mutation screening of the Wolfram syndrome gene in psychiatric patients. Mol Psychiatry 2001; 6: 39–43.
Crawford J, Zielinski MA, Fisher LJ, Sutherland GR, Goldney RD : Is there a relationship between Wolfram syndrome carrier status and suicide? Am J Med Genet 2002; 114: 343–346.
Lesperance MM : The Kresge Hearing Research Institute Human Genetics Laboratory 2004, http://www.khri.med.umich.edu/research/lesperance_lab/lfsnhl.shtml.
Awata T, Inoue K, Kurihara S et al: Missense variations of the gene responsible for Wolfram syndrome (WFS1/wolframin) in Japanese: possible contribution of the Arg456His mutation to type 1 diabetes as a nonautoimmune genetic basis. Biochem Biophys Res Commun 2000; 268: 612–616.
Domenech E, Gomez-Zaera M, Nunes V : WFS1 mutations in Spanish patients with diabetes mellitus and deafness. Eur J Hum Genet 2002; 10: 421–426.
Minton JA, Hattersley AT, Owen K et al: Association studies of genetic variation in the WFS1 gene and type 2 diabetes in UK populations. Diabetes 2002; 51: 1287–1290.
Cryns K, Pfister M, Pennings RJ et al: Mutations in the WFS1 gene that cause low-frequency sensorineural hearing loss are small non-inactivating mutations. Hum Genet 2002; 110: 389–394.
Castro FJ, Barrio J, Perena MF, Palomar MT, Cristobal JA : Uncommon ophthalmologic findings associated with Wolfram syndrome. Acta Ophthalmol Scand 2000; 78: 118–119.
Sumboonnanonda A, Vongjirad A, Suntornpoch V, Angsusingha K, Parichatikanond P, Laohapand T : Renal failure in two patients with Wolfram syndrome. J Pediatr Endocrinol Metab 1997; 10: 645–651.
Bekir NA, Gungor K, Guran S : A DIDMOAD syndrome family with juvenile glaucoma and myopia findings. Acta Ophthalmol Scand 2000; 78: 480–482.
Al-Till M, Jarrah NS, Ajlouni KM : Ophthalmologic findings in fifteen patients with Wolfram syndrome. Eur J Ophthalmol 2002; 2: 84–88.
Sam W, Qin H, Crawford B, Yue D, Yu S : Homozygosity for a 4-bp deletion in a patient with Wolfram syndrome suggesting possible phenotype and genotype correlation. Clin Genet 2001; 59: 136–138.
den Dunnen JT, Antonarakis SE : Nomenclature for the description of human sequence variations. Hum Genet 2001; 109: 121–124.
Kent WJ : BLAT – The BLAST-Like Alignment Tool. Genome Res 2002; 12: 656–664.
Kent WJ, Sugnet CW, Furey TS et al: The Human Genome Browser at UCSC. Genome Res 2002; 12: 996–1006, and UCSC Genome Browser. 2004 (http://genome.ucsc.edu/).
Acknowledgements
The excellent technical assistance in this work was carried out by Lillian Rasmussen, Annemette Mikkelsen and Mona Kristensen, the patients and relatives are thanked for their patience and collaboration. We also thank Henning Laursen, neuro-pathologist at The University Hospital, Rigshospitalet, Copenhagen, for commenting on the autopsia report. The project was hosted by the Wilhelm Johannsen Centre for Functional Genomics and GENOME GROUP/RC-LINK, and supported by a grant from The Danish Society of the Blind, the Oticon Foundation and Widex AS. The Danish National Research Foundation funded The Wilhelm Johannsen Centre for Functional Genomics.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hansen, L., Eiberg, H., Barrett, T. et al. Mutation analysis of the WFS1 gene in seven Danish Wolfram syndrome families; four new mutations identified. Eur J Hum Genet 13, 1275–1284 (2005). https://doi.org/10.1038/sj.ejhg.5201491
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201491
Keywords
This article is cited by
-
A novel mutation of WFS1 gene leading to increase ER stress and cell apoptosis is associated an autosomal dominant form of Wolfram syndrome type 1
BMC Endocrine Disorders (2021)
-
Collaboration for rare diabetes: understanding new treatment options for Wolfram syndrome
Endocrine (2021)
-
Effect of 4-phenylbutyrate and valproate on dominant mutations of WFS1 gene in Wolfram syndrome
Journal of Endocrinological Investigation (2020)
-
Novel mutations and the ophthalmologic characters in Chinese patients with Wolfram Syndrome
Orphanet Journal of Rare Diseases (2019)
-
Wolfram syndrome, a rare neurodegenerative disease: from pathogenesis to future treatment perspectives
Journal of Translational Medicine (2019)
