
Abstract
Three coalescent-based methods allowed us to infer some aspects of the history of three Bulgarian Gypsies populations belonging to the Vlax linguistic group: the Lom, Rudari and Kalderas. We used several kinds of genetic markers: HV1 sequences of the maternally inherited mitochondrial genome and microsatellites of the paternally inherited Y chromosome and of the biparentally inherited chromosome 8. This allowed us to infer several parameters for men and women: the splitting order of the populations and the ages of the splitting events, the growth rate in each population and the migration rates between populations. Altogether, they enabled us to infer a demographic scenario that could explain the genetic diversity of Vlax Roma: recent splits occurring after the arrival in Europe, asymmetric migration flows especially for males and unequal growth rates. This represents a considerable contribution to the Vlax Roma history in comparison with the inferences from classical population genetics.
Similar content being viewed by others
Introduction
The 8–10 million European Roma (Gypsies) are best described as a mosaic of founder populations, the Gypsy groups, with common Asian origins supported by linguistic and cultural,1,2 as well as genetic evidence.3 A recent study of Y chromosome and mitochondrial DNA diversity3 has shown that the current genetic profile of the socially and geographically divergent Gypsy groups has been shaped by their migrations in Europe in the course of the last millennium, with genetic affinities reflecting to a large extent the linguistic classification into Balkan, Vlax and Western European Roma. Each of these large migrational/linguistic categories comprises numerous socially and culturally defined endogamous groups. The Vlax Roma share several centuries of slavery in the Romanian-speaking lands north of the Danube river, the Wallachian Principalities (hence the term ‘Vlax’). Their dispersal to other parts of Europe, and the rest of the world, has occurred in small groups of runaway slaves in the 17th–18th century, and two subsequent large migration waves – in the late 19th–early 20th century – after the abolition of slavery in Romania, and in the late 20th century, following the economic changes in Eastern Europe.
Interest in the genetics of the Vlax Roma has resulted in the identification of several novel single gene disorders and private founder mutations, Hereditary Motor and Sensory Neuropathy types Lom4,5 and Russe,6,7 Congenital Cataracts Facial Dysmorphism Neuropathy Syndrome8,9 and galactokinase deficiency.10,11 A population genetic study of three groups of Vlax Roma12 revealed limited diversity, with a single ancestral Y chromosome lineage shared by 73% of males. The Vlax Roma, who account for a substantial proportion of the overall Gypsy population, thus qualify among the most restricted founder populations of Europe whose potential for the study of genetically complex disorders is still to be explored. At the same time, the possibility of internal differentiation, suggested by the uneven distribution of founder mutations6,8 and by data obtained from the analysis of the highly mutable Y chromosome minisatellite MSY1,12 raises the need for further research into the population history of individual Vlax Roma groups.
In this study, we have used the new approaches based on coalescence theory,13 namely the recently developed programs Migrate,14,15 Fluctuate16,17 and Batwing,18,19 to characterise three Vlax groups in terms of the time of founding through splits from a common ancestral population, subsequent exchange of migrants and historical demography. The use of genetic markers of different mutability from multiple genomic regions, and of either maternal, paternal or biparental inheritance, allowed better parameter estimation and an independent investigation of male and female population history. Estimates were compared to the results of classical population genetics and to the available ethnological, historical and linguistic sources of information.
Subjects and methods
Subjects
The study included a total of 108 males from the Lom, Kalderas and Rudari (previously referred to as Monteni12,3) groups of Vlax Roma. The three groups represent different linguistic branches, with the Lom speaking an ‘old Vlax’ and the Kalderas a ‘new Vlax’ dialect of Romanes, while the Rudari use archaic Romanian (rather than Romanes). The three groups differ markedly in terms of social practices and cultural tradition, and are separated by strict rules of endogamy.12,3 Informed consent has been obtained from all participants in the study.
Genetic markers
Mitochondrial DNA
HVS1 sequencing data on 43 Lom, 42 Rudari and 23 Kalderas were obtained from Kalaydjieva et al12 and Gresham et al.3
Y chromosome
Eight microsatellite loci, namely DYS19, DYS388, DYS389AB, DYS389CD, DYS390, DYS391, DYS393 and DYS392, were genotyped in 45 Lom, 18 Rudari and 11 Kalderas, as reported in Kalaydjieva et al12 and Gresham et al.3 DYS389CD was defined as equivalent to DYS389I and DYS389AB as equivalent to DYS389II minus DYS389I20
Autosomal markers
The analysis included four microsatellites on 8q23 (D8S1835, D8S1708, D8S1746 and D8S1796), surrounding the NDRG1 gene, mutated in Hereditary Motor and Sensory Neuropathy type Lom (HMSNL). Genotyping was as described in Chandler et al.21 Haplotypes were obtained from family studies. Normal, non-transmitted chromosomes were characterised in 31 Lom, 22 Rudari and 21 Kalderas subjects.
Statistical analysis
Classical population genetics methods
These analyses were conducted using Arlequin Version 2001.22
Intrapopulation diversity was assessed using expected heterozygosity H, the number of haplotypes k, the number of polymorphic sites S and the mean number of pairwise differences π.
The genetic structure of the populations was assessed using the AMOVA Φst measure.23 The number of pairwise differences and the Kimura two-parameters distance were employed in the analysis of mitochondrial sequences. For the analysis of microsatellite data, we used the number of different alleles and the sum of squared size differences between haplotypes (with the obtained Φst values denoted Fst and Rst, respectively), the second being the only one to take the presumed evolutionary relationships between alleles into account.
The demographic regime was inferred by using the Tajima,24,25 Ewens-Watterson26 and Fu27 Fs tests of selective neutrality (interpreted as tests of demographic equilibrium in the case of neutral genetic systems), and computing the mismatch distributions28 of mitochondrial sequences. A validity test of the stepwise expansion model based on the mismatch distributions was performed as described.22
Coalescent-based methods
Three coalescent-based programs were used: Migrate14,15 and Fluctuate16,17 from the Lamarc package (http://evolution.genetics.washington.edu), and Batwing18,19 (available at http://www.maths.abdn.ac.uk/~ijw). Details on the programs and the choice of mutation models and search parameters are given in Appendix A. The methods used to estimate effective population size are presented in Appendix B.
Batwing
Y and 8q microsatellite data were analysed using Batwing Version 1.01 to estimate: (a) the population parameter θ for the three populations altogether (2Mu, where u is the mutation rate and M is equal to Ne – the effective size – for a uniparentally inherited gene and to 2Ne for a biparentally inherited gene); (b) the total growth rate; (c) the time to the most recent common ancestor (TMRCA); (d) the parameters of the population ‘supertree’, namely the times of the splitting events, the identity of the populations that split and the proportional size taken up by each population. The program assumes that the populations under study have diverged from an ancestral population at different points in time, have the same growth rate (growth or stationarity can be assumed) and have not exchanged migrants after the splits. Both a constant size and a continuous exponential growth population model were tested. A generational interval of 30 years was assumed.29
Fluctuate
Mitochondrial sequences were analysed with Fluctuate Version 1.1. The program estimates the population parameter θ and the exponential growth rate g (normalised by the mutation rate per site and per generation) in isolated populations. At any time t (in number of generations) in the past, the size of the population is assumed to be N=N0e−gt where N0 is the present population size.
Migrate
Migrate Version 1.2.4 was used to analyse mitochondrial sequences and Y and 8q microsatellites, and assess the parameter θi for each population i and the absolute number of migrants γji from population j to population i (γji=Neimji). It assumes populations of constant size, connected by constant asymmetric migration flows.
In the postanalysis of the output data, we assumed a mutation rate of 10−5 or 10−6 per site per generation for mitochondrial sequences (corresponding respectively, to estimates based on genealogical30,31,32,33 and on phylogenetical data34,35), and of 0.21% for microsatellites.36,37
Results
Intra-population diversity
The Rudari presented substantially lower Y chromosome diversity indices compared to the Lom and the Kalderas. In the Rudari group, the estimated heterozygosity for mitochondrial sequences was also particularly low (0.913), although indices obtained with the infinite sites model (S and π) were higher than those for the Lom and the Kalderas. Diversity analysis based on 8q microsatellites placed the Rudari halfway between the Lom and Kalderas. These results are shown in Table 1.
Inter-population diversity
The observed pattern and intensity of genetic structure was related to the genetic system under examination (Table 2). The three populations were globally differentiated for mitochondrial sequences (Φst=0.037**), but no differentiation was detected between the Lom and the Kalderas. Y chromosome microsatellites revealed some degree of differentiation in AMOVA analyses based on the number of different alleles (Fst=0.061*, Rst=0.118). Differentiation for the Y chromosome markers was evident only in the Lom–Rudari pair (Fst=0.085**). The analysis of chromosome 8 microsatellites showed no global differentiation (Fst=0.021, Rst=0.037), but differentiation was seen in the pairs Lom–Rudari (Fst=0.033*) and Rudari–Kalderas (Fst=0.032*).
Population fissions
Using the Y chromosome data and assuming a constant population size, the time to the most recent common ancestor (MRCA) estimated by Batwing was 17 000 years (95% CI 9800–34 000). Under the assumption of demographic growth, the age of the MRCA was 14 000 years (8300–26 000).
The estimates, based on the Y chromosome data, of the time separating the present generation from the splitting events were identical for the two demographic models (growth and stationarity): 350 years (80–900) for the first split and 100 years (3–450) for the second split. Very similar results were obtained using the 8q microsatellite data, where the first split was estimated at 360 years (40–1400) and the second at 150 years (8–680). While autosomal markers produced larger confidence intervals, the estimated dates of the population fissions were highly congruent.
The Lom Gypsies appear to have split first. Using the Y chromosome data, their separation from the other Vlax Roma was evident in 75% (assuming population growth) to 80% (assuming constant size) of the supertrees represented, when the Markov process reached stationary distribution. The same configuration was observed in 43% of the supertrees generated from the chromosome 8q data for both demographic models.
Size of the founding populations
Using Migrate, the effective female population size was estimated at 300, 300 and 2500, respectively, for the Lom, Rudari and Kalderas. The effective male population size was below 100 for all three populations, with the Rudari having the lowest size.
According to Fluctuate, the population estimator θ was lower for the Rudari (0.040) than for the two other populations (0.076 and 0.104, respectively). The corresponding effective size of the female population obtained with equation (1) (Appendix B) was 1000 (447–2663) for the Lom, 1000 (431–2431) for the Rudari and 800 (272–2227) for the Kalderas, assuming a 10−5/site/generation mutation rate, and 10 times higher with the 10−6/site/generation mutation rate.
The Batwing estimate of θ for the Vlax, using Y chromosome data and a constant size population model, was 1.82 (95% CI 1.14–2.90), with a corresponding effective size of 434 (95% CI 271–691). Under the assumption of demographic growth, θ was 2.28 (95% CI 1.35–4.30) with a corresponding effective size of 305 (95% CI 157–1966).
Demographic growth
The Tajima and Ewens-Watterson statistics did not show any evidence of size variation for any of the three populations (Table 3); however, such evidence was provided by Fu's Fs statistics for the Kalderas (P-value=0.001). The P-value for the Lom sample was also low (0.028), but did not reach the 0.02 P-value corresponding, for the Fs statistics, to a level of significance of 0.05.22
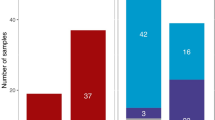
The mismatch distributions (Figure 1) of the Lom and Kalderas mitochondrial sequences were unimodal, with P-values obtained in the test of validity of the stepwise expansion model of 0.13 and 0.14, respectively. By contrast, the distribution was multimodal-like for the Rudari, with a P-value of 0.02. According to these results, the Lom and the Kalderas populations were expanding, whereas a constant size appears to characterise the Rudari.

Mismatch distributions of mitochondrial sequences. The P-values of the validity test of the estimated stepwise expansion model were, respectively, 0.13, 0.02 and 0.14 for the Lom, the Rudari and the Kalderas.
Using Batwing, the absolute growth rate for all three Vlax Roma populations pooled together was 1.0022 per generation (1.00002–1.011).
Using Fluctuate, the estimated exponential growth rate (normalised by mutation rate/site/generation) was lowest for the Rudari (87 (CI 6–168)) and highest for the Kalderas (293 (185–401)), with the Lom Gypsies taking an intermediate position (143 (63–224)). Assuming a mutation rate of 10−5 per site and per generation, this translated into absolute growth rates per generation of 1.0009 (1.00006–1.00168) for the Rudari, 1.0014 (1.00063–1.00224) for the Lom and 1.0030 (1.00185–1.00403) for the Kalderas.
Exchange of migrants
Table 4 presents the mean values of the estimates obtained in several independent runs of Migrate (10 runs for the Y chromosomes, 18 for mitochondrial DNA and seven for chromosome 8q markers). The Y chromosome data provided a clear indication that there was less male immigration than emigration in the Rudari, in terms of both the migration rate m or absolute number of migrants Nm. On the other hand, no clear pattern of migrations emerged from the analyses of mitochondrial DNA and chromosome 8q, where the confidence intervals were largely overlapping. However, these results have to be treated with caution because of the high variability between the estimates provided by the different runs (for the 10 runs performed on the Y chromosomes, the standard deviations of the estimated numbers of migrants ranged from 0.3 to 2.5, which are high values compared to means given in Table 4).
Discussion
Coalescence theory provides the tools for addressing the historical and evolutionary mechanisms resulting in a sample of genes observed today, and thus provides an insight into historical demography.38,39,40 Due to methodological problems and computational intensity, coalescence-based programs have become available only recently,14,15,16,17,18,19 and so far their application to the analysis of demographic parameters has been limited to very few studies.41,42,43 Therefore, before discussing the specific results of this study, it appears useful to comment on the new methods.
The major limitation of coalescent-based approaches is the simplification of the population models: Batwing assumes an absence of migrants between subpopulations, Migrate estimates migration rates between stationary populations and Fluctuate assesses growth rates in isolated populations. Since the history of natural human populations is often more complex, the accuracy of the estimations in those populations is questionable. In our case, coalescent-based methods provided information that was consistent with non-genetic sources. Nevertheless, simulation studies as well as further research into real populations are needed for a rigorous assessment of the sensitivity of these methods to violations of the assumed models.
The three programs used in this study are still under development, and the current limitations do not allow the use of their full potential. The high computational requirements (with a long duration of each run on a powerful UNIX workstation) and high inter-run variability, especially in Migrate, highlight the difficulties related to the use of Monte Carlo Markov chains – the need for a very high number of iterations to obtain convergence44 and the lack of simple criteria for convergence to the maximum-likelihood region. At the time of the study, Batwing and Migrate allowed only for the stepwise mutation model or a similar model in analysing microsatellite data. Although this model is widely used, it would be beneficial to have the possibility of assessing the impact of other mutation models on the estimates.
Notwithstanding the existing limitations, coalescent-based methods increase greatly our ability to estimate demographic parameters. While the classical mismatch distributions and neutrality tests provide a general indication of population growth, Fluctuate allows the assessment of specific growth rates. The moment-based estimators developed to calculate the effective population size and migration rates from one- and two-locus identity measures45 are based on assumptions of equal effective population sizes and symmetrical migration flows. By contrast, Migrate allows complete asymmetry of the system. Furthermore, in our study, the confidence intervals inferred from the coalescent-based approaches were often narrow enough to allow conclusions. In Migrate, the wide confidence intervals of the estimates could be explained by the fact that the 8q and Y loci are partially or totally linked, but the modes of the likelihood curves should not have been affected by this linkage.
The use of the three complementary coalescent-based methods allowed us to outline a demographic scenario that can explain the present genetic structure of the Vlax Roma and the historical relationships between the individual Vlax groups (Figure 2). During most of their history, the Lom, Rudari and Kalderas Gypsies have been part of a single ancestral population. The population splits are very recent in nature, as shown by Batwing, with a very good concordance between the results obtained with autosomal and Y chromosome microsatellites (although one should note that Batwing does not allow for recombinations and the data obtained with the chromosome 8 microsatellites should be treated with caution). Our results are also consistent with the age of the major Y chromosome lineage in the Vlax Roma, estimated previously at 400–500 years.12

Historical demography of the Vlax Roma, as inferred by coalescent-based methods from current genetic diversity.
The reasons behind the split of the original population into separate endogamous groups have been related by social anthropologists to Gypsy slavery in the Wallachian Principalities and Moldova, where the Roma were enslaved almost immediately after their arrival, around the 14th century.46 Depending on the owner, slaves were divided into categories, with further subdivisions based on trade.46 Our data suggest that the formation of the three Romani populations occurred later, in close coincidence with the migrations out of Wallachia. The results indicate that the Lom were the first to separate from the other two populations around 350 years ago, in agreement with the ‘old Vlax’ dialect spoken by this group, suggesting that the Lom derive from the run-away slaves leaving Wallachia in small groups in the 17th–18th centuries. The dating of the second division, between the Kalderas and the Rudari, correlates with the large migration wave after the abolition of slavery, in the late 19th–early 20th century. One should note, however, that the confidence intervals obtained do not allow a definitive distinction between the two scenarios.
The size of the founding populations appears to have been very small, with good agreement between the results produced by the three methods. The total effective population size, estimated by Migrate on the chromosome 8 data, was about 250 for each group. The effective male population size was about 100 for each group according to Migrate, and about 350 for the three groups pooled together according to Batwing. For the effective female size, the estimates provided by Migrate and Fluctuate ranged from 300 to 2500, depending on the population. Information on the historical demography of individual Romani groups is extremely limited, and we can compare the above results only to the 1893 census for Transylvania, where the reported size of individual Rudari and Kalderas groups was in the same range: about 2000 individuals each.47
Demographic growth was assessed using classical as well as coalescent-based methods, with essentially similar results (Figure 1). The total growth rate of 1.0022 per generation (CI 1.00002–1.011), estimated by Batwing on the Y chromosome data, was in the same range as the Fluctuate estimates for the three populations (1.0009–1.0030 per generation). The demographic growth of the Vlax Roma thus appears to have been substantially lower than the estimated rates for the overall European population, namely 1.016 according to Wilson et al19 and 1.027 according to Slatkin and Bertorelle.48 Differences in the historical demography of the three Vlax Roma populations were suggested by the analysis of mtDNA sequence mismatch distributions and by the growth rates estimates by Fluctuate, both characterising the Rudari as a constant-size or very slowly growing population. The growth rates inferred by Batwing and Fluctuate are much lower than the rate previously estimated by Austerlitz et al49 on the same populations (from ∼1.5 to 1.9). However, in this other study, the growth rate was estimated from the analysis of frequency of disease alleles and allelic association with haplotypes surrounding the disease locus. Consequently, only the last 20 generations (the approximate estimated age of the disease alleles) were taken into account, whereas coalescent-based programs infer estimates averaged over the whole history of populations up to the TMRCA (in this case ∼15 000 years old).
The analyses performed with Migrate pointed to an asymmetrical flow of male migrants between the Rudari and the other two populations, with emigration far in excess of immigration. The observed pattern appears consistent with ethnological data. The Rudari emphatically distinguish themselves from all other Gypsy groups, have a preferred Romanian (not Romani) ethnic identity and speak an archaic dialect of Romanian, which has been interpreted as a means to highlight the distinction.50 These attitudes are reflected in marriage patterns: the Rudari are strictly endogamous relative to other Romani groups, but declare the acceptance of intermarriage with non-Romani females.50
The level of pairwise population differentiation (Fst) is consistent with the pattern of splitting events inferred by Batwing from the Y chromosome data: the last split involves Kalderas and Rudari, a pair of populations that show a very limited differentiation for Y chromosome microsatellites. For mitochondrial sequences, there is no clear relation between differentiation and splitting events, because other factors, such as migration flows, might have influenced genetic diversity.
In previous studies,3 we have shown that the population history of the Roma is a string of bottleneck events, with current genetic profiles shaped by differential drift and admixture and carrying the imprint of migrations within Europe. The data that we present here characterise the Lom, Rudari and Kalderas as very young founder populations with a very small historical effective population size and generally slow subsequent demographic growth. This scenario is reflected in the limited genetic diversity, especially of male lineages, shown to be among the most restricted worldwide.12 The smallest historical effective population size, stagnant demographic regime, and an asymmetrical flow of male migrants can explain the extremely limited diversity of the Rudari. These results place the Vlax Roma among the best candidates for research into the genetics of complex disorders. Since the populations split only very recently, it is unlikely that private polymorphism could exist in any of these populations for more stable markers, such as SNP, which are the result of unique mutation events. The data suggest that, despite their social divergence, the individual groups can be treated as a single young founder population, providing access to larger sample sizes of affected subjects and controls.
References
Fraser A : The Gypsies. Oxford: Blackwell Publishers; 1992.
Liégeois J-P : Roma, Gypsies, Travellers. Strasbourg: Council of Europe Press; 1994.
Gresham D, Morar B, Underhill PA et al: Origins and divergence of the Roma (Gypsies). Am J Hum Genet 2001; 69: 1314–1331.
Kalaydjieva L, Hallmayer J, Chandler D et al: Gene mapping in Gypsies identifies a novel demyelinating neuropathy on chromosome 8q24. Nat Genet 1996; 14: 214–217.
Kalaydjieva L, Gresham D, Gooding R et al: N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am J Hum Genet 2000; 67: 47–58.
Rogers T, Chandler D, Angelicheva D et al: A novel locus for autosomal recessive peripheral neuropathy in the EGR2 region on 10q23. Am J Hum Genet 2000; 67: 664–671.
Hantke J, Rogers T, French L et al: Refined mapping of the HMSNR critical gene region – construction of a high-density integrated genetic and physical map. Neuromusc Disord 2003; 13 (9): 729–736.
Angelicheva D, Turnev I, Dye D et al: Congenital cataracts facial dysmorphism neuropathy (CCFDN) syndrome: a novel developmental disorder in Gypsies maps to 18qter. Eur J Hum Genet 1999; 7: 560–566.
Merlini L, Gooding R, Lochmueller W et al: Genetic identity of Marinesco-Sjogren/ myoglobinuria and CCFDN syndromes. Neurology 2002; 58: 231–236.
Kalaydjieva L, Perez-Lezaun A, Angelicheva D et al: A founder mutation in the GK1 gene is responsible for galactokinase deficiency in Roma (Gypsies). Am J Hum Genet 1999; 65: 1299–1307.
Hunter M, Heyer E, Austerlitz F et al: The P28T mutation in the GALK1 gene accounts for galactokinase deficiency in Roma (Gypsy) patients across Europe. Pediatr Res 2002; 51: 602–606.
Kalaydjieva L, Calafell F, Jobling MA et al: Patterns of inter- and intra-group genetic diversity in the Vlax Roma as revealed by Y chromosome and mitochondrial DNA lineages. Eur J Hum Genet 2001; 9: 97–104.
Hey J, Machado CA : The study of structured populations – new hope for a difficult and divided science. Nat Rev Genet 2003; 4: 535–543.
Beerli P, Felsenstein J : Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 1999; 152: 763–773.
Beerli P, Felsenstein J : Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA 2001; 98: 4563–4568.
Kuhner MK, Yamato J, Felsenstein J : Estimating effective population size and mutation rate from sequence data using Metropolis–Hastings sampling. Genetics 1995; 140: 1421–1430.
Kuhner MK, Yamato J, Felsenstein J : Maximum likelihood estimation of population growth rates based on the coalescent. Genetics 1998; 149: 429–434.
Wilson IJ, Balding DJ : Genealogical inference from microsatellite data. Genetics 1998; 150: 499–510.
Wilson IJ, Weale ME, Balding DJ : Inferences from DNA data: population histories, evolutionary processes, and forensic match probabilities. J R Statist Soc A 2003; 166: 1–33.
Rolf B, Meyer E, Brinkmann B, de Knijff P : Polymorphism at the tetranucleotide repeat locus DYS389 in 10 populations reveals strong geographic clustering. Eur J Hum Genet 1998; 6: 583–588.
Chandler D, Angelicheva D, Heather L et al: Hereditary motor and sensory neuropathy – Lom (HMSNL): refined genetic mapping in Romani (Gypsy) families from several European countries. Neuromusc Disord 2000; 10: 584–591.
Schneider S, Roessli D, Excoffier L : Arlequin ver. 2000: a software for population genetics data analysis. Switzerland: Genetics and Biometry Laboratory, University of Geneva; 2000.
Excoffier L, Smouse PE, Quattro JM : Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 1992; 131: 479–491.
Tajima F : Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989; 123: 585–595.
Tajima F : The effect of change in population size on DNA polymorphism. Genetics 1989; 123: 597–601.
Watterson GA : The homozygosity test after a change in population size. Genetics 1986; 112: 899–907.
Fu YX : Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997; 147: 915–925.
Slatkin M, Hudson RR : Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 1991; 129: 555–562.
Tremblay M, Vezina H : New estimates of intergenerational time intervals for the calculation of age and origins of mutations. Am J Hum Genet 2000; 66: 651–658.
Heyer E, Zietkiewicz E, Rochowski A, Yotova V, Puymirat J, Labuda D : Phylogenetic and familial estimates of mitochondrial substitution rates: study of control region mutations in deep-rooting pedigrees. Am J Hum Genet 2001; 69: 1113–1126.
Howell N, Kubacka I, Mackey DA : How rapidly does the human mitochondrial genome evolve? Am J Hum Genet 1996; 59: 501–509.
Parsons TJ, Muniec DS, Sullivan K et al: A high observed substitution rate in the human mitochondrial DNA control region. Nat Genet 1997; 15: 363–368.
Sigurgardottir S, Helgason A, Gulcher JR, Stefansson K, Donnelly P : The mutation rate in the human mtDNA control region. Am J Hum Genet 2000; 66: 1599–1609.
Tamura K, Nei M : Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 1993; 10: 512–526.
Horai S, Hayasaka K, Kondo R, Tsugane K, Takahata N : Recent African origin of modern humans revealed by complete sequences of hominoid mitochondrial DNAs. Proc Natl Acad Sci USA 1995; 92: 532–536.
Heyer E, Puymirat J, Dieltjes P, Bakker E, de Knijff P : Estimating Y chromosome specific microsatellite mutation frequencies using deep rooting pedigrees. Hum Mol Genet 1997; 6: 799–803.
Kayser M, Roewer L, Hedman M et al: Characteristics and frequency of germline mutations at microsatellite loci from the human Y chromosome, as revealed by direct observation in father/son pairs. Am J Hum Genet 2000; 66: 1580–1588.
Kingman : On the genealogy of large populations. J Appl Probab 1982; 19: 27–43.
Tajima F : Evolutionary relationship of DNA sequences in finite populations. Genetics 1983; 105: 437–460.
Fu YX, Li WH : Coalescing into the 21st century: an overview and prospects of coalescent theory. Theor Popul Biol 1999; 56: 1–10.
Harding RM, Fullerton SM, Griffiths RC et al: Archaic African and Asian lineages in the genetic ancestry of modern humans. Am J Hum Genet 1997; 60: 772–789.
Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW : Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc Natl Acad Sci USA 2000; 97: 7360–7365.
Hurles ME, Nicholson J, Bosch E, Renfrew C, Sykes BC, Jobling MA : Y chromosomal evidence for the origins of oceanic-speaking peoples. Genetics 2002; 160: 289–303.
Stephens M, Donnelly P : Inferences in molecular population genetics. J R Statist Soc, Series B 2000; 62: 605–655.
Vitalis R, Couvet D : Estimation of effective population size and migration rate from one- and two-locus identity measures. Genetics 2001; 157: 911–925.
Hancock I : The Pariah Syndrome. Ann Arbor, Michigan: Karoma Publishers Inc.; 1987.
Achim V : Tigani in Istoria Romaniei. Bucuresti: Editura Enciclopedica; 1998.
Slatkin M, Bertorelle G : The use of intraallelic variability for testing neutrality and estimating population growth rate. Genetics 2001; 158: 865–874.
Austerlitz F, Kalaydjieva L, Heyer E : Detecting population growth, selection and inherited fertility from haplotypic data. (in press).
Marushiakova E, Popov V : Gypsies (Roma) in Bulgaria. Studien zur Tsiganologie und Folkloristik. Frankfurt am Main: Editions Peter Lang; 1997.
Excoffier L, Yang Z : Substitution rate variation among sites in mitochondrial hypervariable region I of humans and chimpanzees. Mol Biol Evol 1999; 16: 1357–1368.
Watterson GA : On the number of segregating sites in genetical models without recombination. Theor Popul Biol 1975; 7: 256–276.
Kimura M, Ohta T : Stepwise mutation model and distribution of allelic frequencies in a finite population. Proc Natl Acad Sci USA 1978; 75: 2868–2872.
Abramowitz M, Stegun IA : Handbook of Mathematical Functions, Applied Mathematics Series, Vol. 55, Government Printing Office, Washington DC, 1964.
Acknowledgements
We thank MK Kuhner, P Beerli, IJ Wilson and L Excoffier for their help in the usage of their programs. We thank also Drs A Sibert and B Toupance for helpful comments. This study was funded by the Australian Research Council and The Wellcome Trust. Some runs of the coalescence softwares were performed on the UNIX machine of INRA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg).
Supplementary information
Rights and permissions
About this article
Cite this article
Chaix, R., Austerlitz, F., Morar, B. et al. Vlax Roma history: what do coalescent-based methods tell us?. Eur J Hum Genet 12, 285–292 (2004). https://doi.org/10.1038/sj.ejhg.5201126
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201126
Keywords
This article is cited by
-
Sex-biased patterns shaped the genetic history of Roma
Scientific Reports (2020)
-
Unique frequencies of HFE gene variants in Roma/Gypsies
Journal of Applied Genetics (2012)
-
Hungarian mtDNA population databases from Budapest and the Baranya county Roma
International Journal of Legal Medicine (2007)
