
Abstract
In contrast to the frequent dominant optic atrophies (DOAs) in which the neuropathy is usually an isolated event, isolated recessive optic atrophies (ROAs) are very uncommon and have been described as severe congenital or early infantile conditions. To date, two loci for isolated DOA have been mapped, of which one was ascribed to mutations in the OPA1 gene. Conversely, no isolated autosomal ROA locus had previously been localised. Here, we report a large multiplex consanguineous family of French origin affected with an early onset but slowly progressive form of isolated OA. A genome-wide search for homozygosity allowed the localisation of the disease-causing gene to chromosome 8q21–q22 (Zmax of 3.41 at θ=0 for D8S270), in a 12 Mb interval flanked by markers D8S1702 and D8S1794. This localisation excludes allelism of the disease with both isolated DOAs, on one hand, or all known syndromic forms of ROA, on the other hand, supporting the mapping of a first gene for isolated autosomal ROA (ROA1) on the long arm of chromosome 8.
Similar content being viewed by others


Introduction
Genetically determined optic atrophies (OAs) affect the retinal ganglion cells, the retinal fibre layer or the intraocular portion of the optic nerve. The process may be focal (especially affecting the macular beam of the optic nerve) or generalised.1 Autosomal dominant optic atrophies (DOAs) are the most common form of hereditary optic neuropathy (prevalence 1:50 000). This genetic subtype is characterised by the loss of fibres of the central retina leading to a caecocentral visual field scotoma. Conversely, the peripheral visual field remains strictly normal in advanced stages of the disease. Colour vision is abnormal with frequently a blue–yellow defect. In addition to the long reported intra- and interfamilial clinical variability of DOA, genetic heterogeneity has been demonstrated as two loci have been reported: OPA1 (MIM 165500), by far the most frequent, on chromosome 3q28–q29, and OPA4 (MIM 605293) on chromosome 18q12.2–q12.3. OPA1 has been shown to be accounted for by mutations in the Msp1 protein (MIM 605290).
In sharp contrast to DOA, in which the OA is usually an isolated event, the recessive optic atrophies (ROAs) are often multisystemic diseases involving the central nervous system and other organs. So far, only two genes for syndromic ROA have been identified: OPA3 (19q13.2–q13.3, 3-methylglutaconicaciduria type III, (606580)) and WFS1 (4p16.1, Wolfram syndrome, (MIM 606201)). In addition, one locus for syndromic ROA has been reported: WFS2 on chromosome 4q22–q24 (MIM 604928).
Compared to syndromic ROA, isolated autosomal ROAs are extremely uncommon (MIM 258500).2,3 These cases have been described as congenital or early infantile total OA, that is, with alteration of both macular and peripheral bundles of the optic nerve resulting in the perturbation of both central and peripheral areas of the visual field before the age of 3 years. The visual impairment is severe: nystagmus as well as a severe dyschromatopsia close to achromatopsia are usually noted. A profound alteration in visual acuity is almost always reported. Since their description, these observations have been largely dismissed or overlooked.2,3 In 1992, Moller opened a debate about the existence of these isolated forms of recessively inherited simple OA and concluded that ‘a very clearcut well-documented pedigree has yet to be published’.4
Here, we report a large multiplex consanguineous family segregating true isolated autosomal recessive OA and the mapping of the disease-causing gene on chromosome 8q21–q22 (ROA1).
Materials and methods
Patients
A large multiplex consanguineous family of French origin affected with isolated autosomal recessive OA (Figure 1) was ascertained through the Ophthalmologic Consultation of the ‘Hôpital des Enfants Malades’ of Paris. All family members underwent general and ophthalmologic examinations. The blood of the family members was collected and DNA was purified by phenol–chloroform extraction.

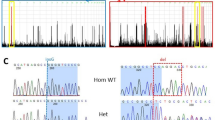
ROA1 family pedigree and haplotype at the 8q21–q22 region. Individuals available for typing are indicated by the generated genotype data. The homozygote haplotype is squared for each patient. One recombination event detected in V2, on one hand, and loss of homozygosity detected in all patients, on the other hand, enabled us to define a critical region between D8S1702 and D8S1794, respectively.
Prior to the present study, the hypothesis of Leber Optic Neuropathy (LHON (MIM 535500)) was excluded by the analysis of the mitochondrial DNA in another laboratory.
Linkage analysis
Four markers containing short tracks of (CA)n repeats flanking the OPA1 (3q28–q29), OPA4 (18q12.2–q12.3) and 3-methylglutaconicaciduria type III (OPA3, 19q13.2–q13.3) loci, as well as the two Wolfram syndrome loci, WFS1 (4p16.1) and WFS2 (4q22–q24), were chosen from the Généthon Linkage Map5 on the basis of their informativity (data not shown, available on request).
A genome-wide search for homozygosity was undertaken with 382 pairs of fluorescent oligonucleotides of the Genescan Linkage Mapping Set, Version II (Perkin-Elmer Cetus) under conditions recommended by the manufacturer. The polymorphic markers have an average spacing of 10 cM throughout the genome. Subsequently, all family members were genotyped for markers AFM244td1, AFM143yh4, AFMa083wg9, MFD185A, AFM165yb10, MFD45A, AFMa226wb5, AFM094vb7, AFM281vd5 and AFMa114xc9 at the D8S525, D8S1702, D8S1838, D8S167, D8S271, D8S88, D8S1724, D8S1818, D8S1794 and D8S1699 loci on chromosome 8q21–q22, respectively. Amplified fragments were electrophoresed and analysed on an automatic sequencer (ABI 3100, Applied Biosystems, Foster City, USA). Linkage analyses were performed using M-LINK and LINKMAP of the 5.1 version of the Linkage program6,7 (gene frequency estimated to 1/1000, penetrance=1). All allele frequencies are available from the CEPH Database.
Screening for mutations of candidate genes
The 18, 10 and 3 exons of the β subunit of the cone photoreceptor cGMP-gated channel gene (CNGB3 (MIM 605080)), the 2,4-dienoyl-coA reductase gene (DECR1 (MIM 222745)) and the pyruvate dehydrogenase phosphatase (PDP) gene, respectively, were amplified using specific primers designed from the intronic sequences close to the intron–exon junctions (not shown, available on request). In addition, the 2 kb promoter sequence upstream of the CNGB3 gene was amplified in four overlapping fragments using specific primers (not shown, available on request). Amplified PCR products were purified by phenol–chloroform extraction, recovered by ethanol precipitation and directly sequenced using the Big Dye Terminator Cycle Sequencing Kit V2 on a 3100 automated sequencer (ABI Prism, Applied Biosystems, Foster City, USA).
Results
Clinical evaluation of isolated ROA family
We report a large multiplex consanguineous family of French origin affected with isolated OA. In this family, 4/5 sibs (two males and two females) were affected with an isolated, early onset but slowly progressive optic neuropathy. All four patients complained, upon examination, of a visual impairment that could not be corrected by glasses commencing between 2 and 6 years of age. At age 10 years, V2 was thoroughly examined. These explorations, including fundus examination, fluorescein angiography, visual field testing, colour vision analysis and electrophysiological recordings, led to the diagnosis of OA without retinal degeneration. The three younger sibs displayed the same phenotype (Figures 2a, b and 3a-c for V4 at age 32 years). Currently, the patients' ages range between 45 and 36 years. For all affected sibs, the progression of the disease was very slow with visual acuity ranging from 1/10 to 2/10 for distant vision. Moderate photophobia and dyschromatopsia with red–green confusion are also noted. None of the four patients were found to have nystagmus. Both parents IV2 and IV3, as well as V1 underwent complete ophthalmologic exploration and no symptom of optic neuropathy was noted. It is worth noting that a normal ocular pressure was measured for all affected and nonaffected members of the family, as well as a normal cup/disc ratio of the optic disk. Neurological examination failed to reveal any developmental failure, pyramidal or extrapyramidal signs, ataxia or hearing loss. Along the same lines, no alopecia, diabetes or any malformation was noted, allowing the exclusion of all known syndromes associated with autosomal ROA.

Visual field and colour vision testings in normal control and patient V4. Results of a normal individual (left column) are compared with those of an affected patient (right column). (a) Dynamic perimetry using a Goldman apparatus to test the visual field. Four isopters are represented (lines of equal retinal sensitivity). Note that the visual field of the patient is almost normal, except for the existence of a relative and partial scotoma at II1. (b) Colour vision was tested using the D-15 Farnsworth's panel. The circling line on the left indicates that the normal individual was able to harmoniously classify the D colour chips 15, while the patient made a disorganised classification with confusion in the red–green axis.

Electroretinogram (ERG) and visual-evoked response (PEV) in patient V4. (a) ERG recordings showing a normal retinal response for red, blue and white light-components, respectively. (b) Flash visually evoked response: the responses are detectable but display atypical morphology for white, red and blue light components, respectively. (c) VER derived from alternating checkerboard: the responses are badly detectable. Note the reduced amplitude measurements for the 15, 30, and 60, in check size, respectively.
The four patients of the family were born to healthy second cousins (Figure 1). This consanguinity associated with (i) the existence of a common affected ancestor, (ii) the absence of any affected individual in the common branch, ruling out autosomal dominant inheritance with incomplete penetrance, (iii) the transmission of the disease gene through four healthy men (II2, III2, III3 and IV3), ruling out X-linked inheritance and (iv) the affected male–female ratio of 1, strongly support the conclusion of autosomal recessive inheritance.
Primary mapping of the disease-causing gene
After exclusion of the OPA1, OPA4 and 3-methylglutaconicaciduria type III (OPA3) loci on chromosome 3q28–q29, 18q12.2–q12. 3 and 19q13.2–q13.3, respectively, as well as the two Wolfram syndrome loci, WFS1 and WFS2 on chromosome 4p16.1 and 4q22–q24, respectively, a 10-cM genome-wide search for homozygosity was performed to identify the genetic locus associated with the disease. The genome-wide search in this multiplex family showed only one chromosomal region in which informative markers were found to be homozygous in all four patients. Indeed, all patients shared a common homozygote haplotype on chromosome 8q21–q22. One recombination event detected in V2, on one hand, and loss of homozygosity detected in all patients, on the other hand, enabled us to define a critical region between D8S1702 and D8S1794, respectively (Figure 1). The maximum lod score of 3.41 was obtained at θ=0 with the marker D8S270 (Table 1). According to the UCSC Human Genome Project Working Draft database (April 2003 release), the flanking markers D8S1702 and D8S1794 span a 12 Mb candidate region, which contains, to date, 25 known genes and >100 predicted genes.
On the other hand, five other markers were found to be homozygote in three among the four affected sibs (parents fully informative; D6S309, D6S292, D15S130, D18S478 and D22S420). Pairwise lod scores were calculated and shown on Table 2. These lod scores cannot totally exclude linkage with the regions of interest, but for 2/5 markers, the healthy brother was homozygote and genoidentical to his affected sibs (D6S309 and D22S420; data not shown). In addition, flanking markers have been systemically studied for each of the five regions and showed recombination events in affected sibs.
Candidate genes study
To date, 3/25 known genes, that is, the β subunit of the cone photoreceptor cGMP gated channel encoding gene (CNGB3), the 2,4-dienoyl-coA reductase encoding gene (DECR1) and the gene for PDP, have been screened in one patient of the family by direct sequencing using intronic primers flanking each exon, as well as the 2 kb promoter of CNGB3, but no mutations have been found.
Discussion
The existence of isolated ROA has long been discussed. In 1992, Moller reviewed 22 ‘old’ papers suggesting the existence of a recessive form of simple OA. In particular, he pointed out that in 1967, Fraser and Friedmann8 found no such cases in their classic survey on causes of blindness in children. Likewise, he called to mind the review over 18 years of registration of visually impaired Danish children showing no case of isolated ROA. Thus, Moller4 concluded that a clearcut, well-documented pedigree had yet to be published. We describe here a family with an unambiguous isolated autosomal recessive form of optic neuropathy, thus replying to the controversy raised by Moller. It is worth noting that, in contrast to the few families formerly reported with a very early and severe form of isolated autosomal recessive OA, in this pedigree, the four patients are affected with an early onset but moderately progressive form of the disease. The clinical phenotype is strikingly different from the dominant forms (absence of central scotoma and red–green dyschromatopsia).
Considering that numerous genes have been found to be involved both in autosomal recessive and dominant diseases (eg: Rho,9,10 GUCY2D,11,12 CRX,13,14 etc), the two already known DOA loci (OPA1, OPA4) were studied in this family by linkage analysis and excluded. Subsequently, allelism with autosomal recessive syndromic OAs was also considered but no linkage was found for the OPA3, WFS1 and WFS2 loci.
Consequently, a genome-wide search for homozygosity was undertaken and a single homozygous region on the long arm of chromosome 8 (8q21–q22) found. It is true that for five markers spared throughout the genome, homozygosity was found in 3/4 affected sibs. Nevertheless, for each region, flanking markers have been studied and showed recombination events in affected sibs. In addition, it is worth noting that for two markers (D6S309 and D22S420), the healthy brother was homozygote and geno-identical to his affected sibs. Moreover, for markers D15S130 and D18S478, the homozygote allele has a frequency in the general population >0.50. Although the lod scores are not <−2, these results are not in favour of linkage.
The absence of any other common homozygous region in all patients excluded allelism between ROA1 and additional known loci of syndromic forms of ROA, not studied at first (these syndromic ROA can be found in the Online Mendelian Inheritance in Man under the query ‘OA’). Convincing lod scores were obtained for several markers covering the genetic interval at 8q21–q22. It is worth noting that the variance of maximal lod score values at θ=0 only reflects the informativity of markers used.
The 12 Mb physical interval between D8S1702 and D8S1794 contains numerous genes of interest. Among them, CNGB3 was first considered. Indeed, although the phenotype of all patients was strikingly different from that described in total colour blindness, the report of severe dyschromatopsia close to achromatopsia described in the few known cases of isolated autosomal recessive OA2,3 prompted us to search for mutations in this gene; however, no mutation was identified allowing the exclusion of this gene.
It is worth remembering that all known genes involved in hereditary optic neuropathies are either mitochondrial (LHON) or encode proteins with high mitochondrial targeting (OPA1, OPA3, TIMM8A (MIM 300356) and SDHA15). This is why we first decided to screen genes known to encode mitochondrial protein such as DECR1 and PDP. The absence of any mutation in these genes prompts us to search in the genetic interval defined by D8S1702 and D8S1794 all proteins potentially targeted to the mitochondria, using the prediction programs MITOPROT and PSORT II from the Mitochondria Project and PSORT server, respectively. This work is currently in progress.
Further studies are required to identify the causative ROA gene in this family and to delineate the role of this locus in small nonconsanguineous families affected with ROA in which linkage analysis cannot be conclusive. The identification of genes involved in ROA will allow the dissection of molecular mechanisms underlying optic neuropathies to aid genetic counselling and to develop rational therapeutic tools.
References
Kaplan J, Rozet JM : Encyclopedia of the human genome; in Macmillan Publishers Ltd, Nature Publishing Group. Hereditary visual disorders, Chapter 510 2003, pp 1–12.
François J : Affections du nerf optique; in Masson (eds). L'hérédité en ophtalmologie. Paris, 1958, pp 581–606.
Merin S : Inherited diseases of the optic nerve; in Dekker M (eds). Inherited eye diseases, diagnosis and clinical management. New York, 1991, pp 323–344.
Moller HU : Recessively inherited, simple optic atrophy – does it exist [Letter]. Ophthal Paediatr Genet 1992; 13: 31–32.
Dib C, Faure S, Fizames C et al: A comprehensive genetic map of the human genome based on 5264 microsatellites. Nature 1996; 380: 152–154.
Lathrop GM, Lalouel JM : Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 1984; 36: 460–465.
Lander ES, Botstein D : Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 1987; 236: 1567–1570.
Fraser GR, Friedmann AI : The causes of blindness in childhood. A study of 776 children with severe visual handicaps. Baltimore, MD, Johns Hopkins Press, 1967.
Dryja TP, McGee TL, Hahn LB et al: Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N 1990; 323: 1302–1307.
Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP : A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet 1992; 1: 209–213.
Perrault I, Rozet JM, Calvas P et al: Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat Genet 1996; 14: 461–464.
Kelsell RE, Gregory-Evans K, Payne AM et al: Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone–rod dystrophy. Hum Mol Genet 1998; 7: 1179–1184.
Freund CL, Gregory-Evans CY, Furukawa T et al: Cone–rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997; 91: 543–553.
Freund CL, Wang Q-L, Chen S et al: De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis? [Letter]. Nat Genet 1998; 18: 311–312.
Birch-Machin MA, Taylor RW, Cochran B et al: Late-onset optic atrophy, ataxia, and myopathy associated with a mutation of a complex II gene. Ann Neurol 2000; 48: 330–335.
Acknowledgements
This work was supported by the Association Retina France and the Association Valentin Haüy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Electronic Database Information
CEPH Database, http://www.cephb.fr/test/cephdb/ (for identification of allelic frequencies).
Mitochondria Project, Database for mitochondria-related genes, proteins and disease, http://www.mips.biochem.mpg.de/proj/medgen/mitop/
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/ (for identification of candidate genes in locus of interest).
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM (for OPA1 (MIM 165500, 605290), OPA4 (MIM 605293), OPA3 (MIM 606280), WFS1 (MIM 606201), WFS2 (MIM 604928), Optic Atrophy Congenital or Early Infantile (MIM 258500), LHON (MIM 535000), CNGB3 (MIM 605080), DECR1 (MIM 222745), TIMM8A (MIM 300356)).
PSORT, Prediction of Protein Sorting Signals and Localization Sites in Amino Acid Sequences, http://psort.nibb.ac.jp/
UCSC Human Genome Project Working Draft, http://genome.ucsc.edu (for marker order and genetic distance between markers).
Rights and permissions
About this article
Cite this article
Barbet, F., Gerber, S., Hakiki, S. et al. A first locus for isolated autosomal recessive optic atrophy (ROA1) maps to chromosome 8q. Eur J Hum Genet 11, 966–971 (2003). https://doi.org/10.1038/sj.ejhg.5201070
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201070
Keywords
This article is cited by
-
Clinical and genetic spectrums of 413 North African families with inherited retinal dystrophies and optic neuropathies
Orphanet Journal of Rare Diseases (2022)
-
Hereditäre Optikusneuropathien
Der Ophthalmologe (2009)
-
Molecular genetic basis of primary inherited optic neuropathies
Eye (2004)
