
Abstract
The COCH gene is the only gene identified in man that causes autosomal dominantly inherited hearing loss associated with vestibular dysfunction. The condition is rare and only five mutations have been reported worldwide. All affected families showed a similar progressive hearing loss and vestibular dysfunction. Since Meniere's disease-like symptoms have also been described in some families, it was suggested that COCH mutations might be present in some patients diagnosed with Meniere's disease. In this study, using a Japanese population, we performed a COCH mutation analysis in 23 patients from independent families with autosomal dominant hearing impairment, four of whom reported vestibular symptoms, and also in 20 Meniere's patients. While a new point mutation, A119 T, was found in a patient with autosomal dominant hearing loss and vestibular symptoms, no mutations were found in the Meniere's patients. Like all other previously identified COCH mutations, the mutation identified here is a missense mutation located in the FCH domain of the protein. The current mutation is located in close spatial proximity to W117, in which a mutation (W117R) had previously been associated with autosomal dominant hearing loss. Model building suggests that, like the W117R mutation, the A119 T mutation does not affect the structural integrity of the FCH domain, but may interfere with the interaction with a yet unknown binding partner. We conclude that mutations in the COCH gene are responsible for a significant fraction of patients with autosomal dominantly inherited hearing loss accompanied by vestibular symptoms, but not for dominant hearing loss without vestibular dysfunction, or sporadic Meniere's disease.
Similar content being viewed by others


Introduction
The COCH gene is known to be responsible for non-syndromic autosomal dominant hearing loss parallelled by vestibular symptoms (DFNA9).1 To date, five COCH mutations have been reported in families of European ancestry.1,2,3,4,5 (Table 1) These include three private mutations in families from the US, a single founder mutation in many families in Belgium and The Netherlands, and a private Australian mutation. For some mutations, the penetrance of vestibular symptoms is complete (the Belgian–Dutch P51S mutation and the Australian I109N mutation). For the US mutations (V66G, G88E, W117R), it was unclear whether the vestibular phenotype was present in all carriers of the COCH mutation. It is therefore not excluded that the penetrance of the vestibular symptoms varies across families, according to the mutation.
Fransen et al3 and Verstreken et al6 reported a large DFNA9 family where, apart from hearing loss and vestibular dysfunction, 25% of the carriers of the COCH mutation suffered from recurrent episodes of dizziness, associated with tinnitus, aural fullness, nausea, and vomiting. Such episodes are very similar to the vertigo attacks encountered in Meniere's disease. Meniere's disease is a term generally used to describe patients with disabling episodes of combined vestibular and auditory disturbance. Both familial and sporadic forms occur. The etiology of this frequent pathology is not known, but it is probably complex, involving multiple environmental causes and predisposing genes.
Due to the similarity between DFNA9 and Meniere's disease, Fransen et al3 suggested that the possibility of a COCH mutation should be considered in patients diagnosed with Meniere's disease. However, there are also clear clinical differences between these two conditions. In Meniere's disease, the hearing loss that accompanies the vertigo attack is usually unilateral and fluctuating, affecting mainly the low frequencies at an early stage of the disease, whereas DFNA9 is characterized by bilateral and progressive hearing loss, starting in the high frequencies and gradually affecting all frequencies.
The biological function of COCH is unclear. COCH mRNA is known to be predominantly expressed in the inner ear7,8 and recent proteomic analysis indicated that the COCH protein, cochlin, constitutes 70% of all inner ear proteins,9 suggesting that this protein plays an important role in the inner ear. The human COCH gene contains two different types of domains, a region homologous to a domain in factor C of Limulus (FCH, factor C homologous domain; sometimes also referred to as LCCL domain)10 and two von Willebrand factor A-like domains (vWFA1 and 2). Interestingly, all mutations reported so far were detected in the FCH domain.
Families with autosomal dominant cochleo-vestibular dysfunction are rare compared to those with autosomal dominant hearing impairment without vestibular symptoms. In addition, both these conditions are very rare in comparison with Meniere's disease. To analyze the contribution of COCH mutations to various forms of hearing and vestibular dysfunction in the Japanese population, we performed COCH mutation analysis in patients with autosomal dominant hearing loss with and without vestibular symptoms, as well as in patients with sporadic Meniere's disease.
Subjects and methods
Subjects
In the present study, we used individuals with Japanese ancestry. All patients and family members gave informed consent to be included in this study. A blood sample was obtained from each individual and DNA was isolated using standard procedures.
Autosomal dominant families
We analyzed 23 subjects from independent families with probable autosomal dominant sensorineural hearing loss (with two or more generations affected). The number of patients in each family ranged from 3 to 65, with a median of 29.2. The severity of hearing loss (a three-frequency average of 500, 1000, 2000 Hz) varied from mild to profound. The hearing loss was mild (30–49 dB) in six patients (15%), moderate (50–69 dB) in nine (40%), severe (70–89 dB) in two (5%), and profound (>90 dB) in six (15%). The age of onset varied from congenital to 40 years. In seven patients (30.4%) the hearing loss was progressive and four patients (17.4%) complained of vertigo.
Meniere's disease patients
Diagnostic criteria for 20 Meniere's patients were according to the guidelines of the American Academy of Otolaryngology-Head and Neck Surgery.11 In brief, the patients were selected by the following clinical features: (1) two or more definitive spontaneous episodes of vertigo lasting 20 min or longer; (2) audiometrically documented hearing loss on at least one occasion; (3) tinnitus or aural fullness. The age of the Meniere's disease patients ranged from 33 to 63 years (mean 50.15). None of the Meniere's disease patients had a family history.
Clinical examination
Pure-tone audiometry was performed in all the patients. The criteria used to consider an individual affected was bilateral sensorineural hearing loss of more than 30 dB in at least one frequency with pure-tone audiometry. For patients with a history of vertigo, a caloric test was performed. CT examination was performed for all patients, but no malformations of the inner ear were identified.
Mutation analysis of the COCH gene
All 12 COCH exons were PCR amplified using previously described intronic primers.1 Amplification products were purified using QIAquick (Qiagen) or gel purified by electroelution, and sequenced using a Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems). Fragments were analyzed using an ABI 377 automated sequencer.
Structure analysis
The mutation found in the present study was modelled onto the nuclear magnetic resonance (NMR) structure of the FCH domain.12 The program MOLMOL13 was used for manual, interactive modelling and to plot Figure 1.

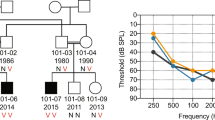
Pedigree of the family bearing a COCH mutation. Filled symbols represent affected members.
Results
A mutation analysis was performed by genomic DNA sequencing of all 12 COCH exons in 23 patients with autosomal dominant hearing loss and in 20 patients with Meniere's disease. No mutations were found in the Meniere's disease patients. A novel COCH mutation was found in one affected member of a family that had affected subjects in two generations (Figure 1). The proband visited our ENT clinic at the age of 47 years because of recurrent dizziness/vertigo and gradually progressive hearing loss associated with tinnitus that had started at the age of 35 years. Pure-tone audiograms showed profound deafness for all frequencies. Caloric testing showed bilateral decreased response. No other obvious clinical abnormalities were noted. Anamnestically, the deceased mother of the proband also had suffered from hearing loss and recurrent episodes of vertigo, starting in her 30s.
The novel COCH mutation detected in this Japanese family is a heterozygous 355A>G mutation in exon 5, resulting in an alanine to threonine substitution at residue 119 of the COCH protein (A119 T). This alanine residue is evolutionarily conserved in the mouse Coch gene, but not in chicken. This mutation was not found in unaffected members in the family (Figure 1; III–1,2,3,4). To exclude that the A119 T variation is a common polymorphism, 96 unrelated random individuals of Japanese origin were analyzed. None of them showed this mutation. No other DNA variations that would lead to amino-acid changes or could affect splice sites were found.
Inspection of the three-dimensional structure of the FCH domain showed that the side chain of A119 is quite solvent-exposed. Using a χ1 angle of −60°, a Thr side chain could be modelled at the site of this residue without introduction of steric clashes in the existing structure (Figure 2).

Stereo representation of a model of the FCH domain with alanine at position 119 replaced by threonine, based on the conformer closest to the mean conformer of the NMR structure. All bonds connecting heavy atoms are shown. The side chain of threonine at position 119 is drawn in a ball-and-stick representation, with red and gray spheres identifying the location of the side-chain oxygen and methyl carbon, respectively. The side chain of tryptophan at position 117 is highlighted with bold lines. Backbone bonds are drawn in magenta. Side chains are drawn in yellow for hydrophobic residues (Ala, Cys, Ile, Leu, Met, Phe, Pro, Trp, Val), gray for uncharged hydrophilic residues (Asn, Gln, Ser, Thr, Tyr), red for negatively charged residues (Asp, Glu), and blue for positively charged residues (Arg, His, Lys). As residue 119 is located on the surface of the domain and its side chain is solvent exposed, the A119T mutation is unlikely to interfere with the structural integrity of the FCH domain.
Discussion
In this study, we found a novel COCH mutation, A119 T, in a Japanese pedigree with hereditary sensorineural hearing loss associated with vestibular impairment. Although only one affected family member was genotyped, the identified mutation is most likely disease-causing. A119 T is not a common variation in the COCH gene since it was not found in unaffected family members or controls, nor has it ever been reported in any other mutation analysis of the COCH gene. The clinical picture of the Japanese patient is highly similar to previously reported patients with a COCH mutation,6 and the disease was likely familial with a segregation pattern compatible with autosomal dominant inheritance.
Previously, five COCH mutations have been reported (P51S, V66G, G88E, I109N, W117R); all in families with European ancestry.1,2,3 (Table 1) A119 T- is the first COCH mutation identified in a patient of nonEuropean ancestry. In Japanese patients, MYO714, KCNQ415, and TECTA16 already have been reported as responsible for nonsyndromic autosomal dominant sensorineural hearing loss. This study adds COCH as a fourth gene.
Including the present mutation, all six mutations in the COCH gene were missense mutations located in the conserved cysteine-rich factor C homologous (FCH) domain (Table 1). This suggests an important specific role for this domain in the functioning of COCH in the inner ear.
The three-dimensional solution structure of this domain has recently been reported.12 It assumes a unique fold that is affected by the majority of the previously reported point mutations. Among the reported mutations, P51S, V66G, G88E, and I109N result in misfolding of the FCH domain. The W117R mutation, however, is correctly folded and is stable as the wild-type protein. This residue is consequently thought to participate in the interaction with a binding partner.12 Also, the A119 T mutation is unlikely to interfere with the structural integrity of the FCH domain. Interestingly, the side chains of A119 and W117 are spatially close (Figure 1), and it is therefore possible that these two mutations interfere with the same process.
Histological analysis of temporal bones of patients with a COCH mutation revealed acidophilic deposits, described as a mucopolysaccharide-like substance, in the cochlea and vestibular end organs.17,18,19 In situ hybridization and immunohistochemical analysis showed expression of COCH in the fibrocytes of the spiral ligament, the spiral limbus, osseous spiral lamina, and the stroma of the vestibular end organs. The deposits were found at exactly the same places in temporal bone sections.8 It was therefore suggested that mutant protein may have a direct or indirect effect on these fibrocytes and lead to degeneration. This may have a negative impact on the inner ear, possibly by interfering with K+ recycling, which is thought to play an important role in ion homeostasis.
The clinical features found in the Japanese patient with the COCH mutation, who showed progressive sensorineural hearing loss starting in the fourth decade of life in combination with recurrent vertigo attacks, are in line with the previous reports of DFNA9 families. The sensorineural hearing loss caused by COCH mutations initially affects only the high frequencies, but progresses to include the lower frequencies, leading to severe to profound loss across all frequencies. The onset is in adulthood, ranging from the third to the fifth decade, and penetrance of the hearing loss is complete. Meniere's disease-like symptoms such as vertigo attacks have only been reported in patients from the Belgian–Dutch families, the present patient, and in a few patients from the Australian family.
No mutations were detected in our 20 sporadic patients diagnosed with Meniere's disease, suggesting that COCH mutations may not be a major cause for typical Meniere's disease, at least not for the sporadic form. Among the 23 patients originating from autosomal dominant families analyzed in this study, four had vertigo complaints and the A119 T mutation was found in one of these four. No mutations were found in any of the autosomal dominant families without vestibular complaints. The absence of mutations in the hearing loss families without vestibular symptoms and in Meniere's disease patients indicates that COCH mutation screening has little chance of success in these patients, and is best limited to autosomal dominant families with adult onset combined hearing loss and vestibular dysfunction.
References
Robertson NG, Lu L, Heller S et al: Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat Genet 1998; 20: 299–303.
de Kok YJ, Bom SJ, Brunt TM et al: A Pro51Ser mutation in the COCH gene is associated with late onset autosomal dominant progressive sensorineural hearing loss with vestibular defects. Hum Mol Genet 1999; 8: 361–366.
Fransen E, Verstreken M, Verhagen WI et al: High prevalence of symptoms of Meniere's disease in three families with a mutation in the COCH gene. Hum Mol Genet 1999; 8: 1425–1429.
Fransen E, Verstreken M, Bom SJ et al: A common ancestor for COCH related cochleovestibular (DFNA9) patients in Belgium and The Netherlands bearing the P51S mutation. J Med Genet 2001; 38: 61–65.
Kamarinos M, McGill J, Lynch M, Dahl H : Identification of a novel COCH mutation, I109N, highlights the similar clinical features observed in DFNA9 families. Hum Mutat 2001; 17: 351.
Verstreken M, Declau F, Wuyts FL et al: Hereditary otovestibular dysfunction and Meniere's disease in a large Belgian family is caused by a missense mutation in the COCH gene. Otol Neurotol 2001; 22: 874–881.
Robertson NG, Skvorak AB, Yin Y et al: Mapping and characterization of a novel cochlear gene in human and in mouse: a positional candidate gene for a deafness disorder, DFNA9. Genomics 1997; 46: 345–354.
Robertson NG, Resendes BL, Lin JS et al: Inner ear localization of mRNA and protein products of COCH, mutated in the sensorineural deafness and vestibular disorder, DFNA9. Hum Mol Genet 2001; 10: 2493–2500.
Ikezono T, Omori A, Ichinose S, Pawankar R, Watanabe A, Yagi T: Identification of the protein product of the Coch gene (hereditary deafness gene) as the major component of bovine inner ear protein. Biochim Biophys Acta 2001; 1535: 258–265.
Trexler M, Banyai L : Patthy L: The LCCL module. Eur J Biochem 2000; 267: 5751–5757.
Committee on Hearing and Equilibrium guidelines for the diagnosis and evaluation of therapy in Meniere's disease. American Academy of Otolaryngology-Head and Neck Foundation, Inc. Otolaryngol Head Neck Surg 1995; 113: 181–185.
Liepinsh E, Trexler M, Kaikkonen A et al: NMR structure of the LCCL domain and implications for DFNA9 deafness disorder. Embo J 2001; 20: 5347–5353.
Koradi R, Billeter M, Wuthrich K : MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 1996; 14: 51–55, 29–32.
Liu XZ, Walsh J, Tamagawa Y et al: Autosomal dominant non-syndromoic deafness (DFNA11) caused by a mutation in the myosin VIIA gene. Nat Genet 1997b; 17: 268.
Akita J, Abe S, Shinkawa H, Kimberling WJ, Usami S : Clinical and genetic features of non-syndromic autosomal dominant sensorineural hearing loss: KCNQ4 is a responsible gene in Japanese. J Hum Genet 2001; 46: 355–361.
Iwasaki S, Harada D, Usami S, Nagura M, Takeshita T, Hoshino T : Association of clinical features with mutation of TECTA in a family with autosomal dominant hearing loss. Arch Otolaryngol Head Neck Surg 2002; 128: 913–917.
Khetarpal U, Schuknecht HF, Gacek RR, Holmes LB : Autosomal dominant sensorineural hearing loss: pedigrees, audiologic findings, and temporal bone findings in two kindreds. Arch Otolaryngol Head Neck Surg 1991; 117: 1032–1042.
Khetarpal U : Autosomal dominant sensorineural hearing loss: further temporal bone findings. Arch Otolaryngol Head Neck Surg 1993; 119: 106–108.
Merchant SN, Linthicum FH, Nadol Jr JB : Histopathology of the inner ear in DFNA9. Adv Otorhinolaryngol 2000; 56: 212–217.
Acknowledgements
We thank the families who participated in the present project. We also thank Ms C Kawashima for technical assistance and Ms AC Apple-Mathews for help in preparing the manuscript. This work is supported by the Ministry of Health and Welfare, Japan (SU), a Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan (SU), the University of Antwerp (GVC), and the Flemish fund for Scientific Research (EF) (GVC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Usami, Si., Takahashi, K., Yuge, I. et al. Mutations in the COCH gene are a frequent cause of autosomal dominant progressive cochleo-vestibular dysfunction, but not of Meniere's disease. Eur J Hum Genet 11, 744–748 (2003). https://doi.org/10.1038/sj.ejhg.5201043
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201043
