
Abstract
Bacillus subtilis is the best-characterized member of the Gram-positive bacteria. Its genome of 4,214,810 base pairs comprises 4,100 protein-coding genes. Of these protein-coding genes, 53% are represented once, while a quarter of the genome corresponds to several gene families that have been greatly expanded by gene duplication, the largest family containing 77 putative ATP-binding transport proteins. In addition, a large proportion of the genetic capacity is devoted to the utilization of a variety of carbon sources, including many plant-derived molecules. The identification of five signal peptidase genes, as well as several genes for components of the secretion apparatus, is important given the capacity of Bacillus strains to secrete large amounts of industrially important enzymes. Many of the genes are involved in the synthesis of secondary metabolites, including antibiotics, that are more typically associated with Streptomyces species. The genome contains at least ten prophages or remnants of prophages, indicating that bacteriophage infection has played an important evolutionary role in horizontal gene transfer, in particular in the propagation of bacterial pathogenesis.
Similar content being viewed by others


Main
Techniques for large-scale DNA sequencing have brought about a revolution in our perception of genomes. Together with our understanding of intermediary metabolism, it is now realistic to envisage a time when it should be possible to provide an extensive chemical definition of many living organisms. During the past couple of years, the genome sequences of Haemophilus influenzae, Mycoplasma genitalium, Synechocystis PCC6803, Methanococcus jannaschii, M. pneumoniae, Escherichia coli, Helicobacter pylori, Archaeoglobus fulgidus and the yeast Saccharomyces cerevisiae have been published in their entirety1,2,3,4,5,6,7,8, and at least 40 prokaryotic genomes are currently being sequenced. Regularly updated lists of genome sequencing projects are available at http://www.mcs.anl.gov/home/gaasterl/genomes.html(Argonne National Laboratory, Illinois, USA) and http://www.tigr.org(TIGR, Rockville, Maryland, USA).
The list of sequenced microorganisms does not currently include a paradigm for Gram-positive bacteria, which are known to be important for the environment, medicine and industry. Bacillus subtilis has been chosen to fill this gap9,10 as its biochemistry, physiology and genetics have been studied intensely for more than 40 years. B. subtilis is an aerobic, endospore-forming, rod-shaped bacterium commonly found in soil, water sources and in association with plants. B. subtilis and its close relatives are an important source of industrial enzymes (such as amylases and proteases), and much of the commercial interest in these bacteria arises from their capacity to secrete these enzymes at gram per litre concentrations. It has therefore been used for the study of protein secretion and for development as a host for the production of heterologous proteins11. B. subtilis (natto) is also used in the production of Natto, a traditional Japanese dish of fermented soya beans.
Under conditions of nutritional starvation, B. subtilis stops growing and initiates responses to restore growth by increasing metabolic diversity. These responses include the induction of motility and chemotaxis, and the production of macromolecular hydrolases (proteases and carbohydrases) and antibiotics. If these responses fail to re-establish growth, the cells are induced to form chemically, irradiation- and desiccation-resistant endospores. Sporulation involves a perturbation of the normal cell cycle and the differentiation of a binucleate cell into two cell types. The division of the cell into a smaller forespore and a larger mother cell, each with an entire copy of the chromosome, is the first morphological indication of sporulation. The former is engulfed by the latter and differential expression of their respective genomes, coupled to a complex network of interconnected regulatory pathways and developmental checkpoints, culminates in the programmed death and lysis of the mother cell and release of the mature spore12. In an alternative developmental process, B. subtilis is also able to differentiate into a physiological state, the competent state, that allows it to undergo genetic transformation13.
General features of the DNA sequence
Analysis at the replicon level.The B. subtilis chromosome has 4,214,810 base pairs (bp), with the origin of replication coinciding with the base numbering start point14, and the terminus at about 2,017 kilobases (kb)15. The average G+C ratio is 43.5%, but it varies considerably throughout the chromosome. This average is also different if one considers the nucleotide content of coding sequences, for which G and A (24% and 30%) are relatively more abundant than their counterparts C and T (20% and 26%). A significant inversion of the relative G− C/G + C ratio is visible at the origin of replication, indicating asymmetry of the nucleotide composition between the replication leading strand and the lagging strand16. Several A+ T-rich islands are likely to reveal the signature of bacteriophage lysogens or other inserted elements (Fig. 1, see below).

Location of genes from class 3 according to codon usage analysis (see Fig. 4) is indicated by dots at the bottom of the graph. Known prophages (PBSX, SPβ and skin) are indicated by their names, and prophage-like elements are numbered from 1 to 7.
We have analysed the abundance of oligonucleotides (‘words’) in the genome in various ways: absolute number of words in the genomic text, or comparison with the expected count derived from several models of the chromosome (for example, Markov models, or simulated sequences in which previously known features of the genome were conserved17). Comparing the experimental data with various models allowed us to define under- and overrepresentation of words in the experimental data set by reference to the model chosen. In general, the dinucleotide bias follows closely what has been described for other prokaryotes18,19, in that the dinucleotides most overrepresented are AA, TT and GC, whereas those less represented are TA, AC and GT. Plots of the frequencies of AG, GA, CT and TC in sliding windows along the chromosome show dramatic decreases or increases around the origin and terminus of replication (data not shown). Trinucleotide frequency, directly related to the coding frame, will be discussed below. The distribution of words of four, five and six nucleotides shows significant correlations between the usage of some words and replication (several such oligonucleotides are very significantly overrepresented in one of the strands and underrepresented in the other one).
Setting a statistical cut-off for the significance of duplications at 10−3, we expected duplication by chance of words longer than 24 nucleotides to be rare20. In fact, the genome of B. subtilis contains a plethora of such duplications, some of them appearing more than twice. Among the duplications, we identified, as expected, the ribosomal RNA genes and their flanking regions, but also regions known to correspond to genes comprising long sequence repeats (such as pks and srf). We also found several regions that were not expected: a 182-bp repetition within the yyaL and yyaO genes; a 410-bp repetition between the yxaK and yxaL genes; an internal duplication of 174 bp inside ydcI; and significant duplications in the regions involved in the transcriptional control of several genes (such as 118 bp repeated three times between yxbB and yxbC). Finally, we found several repetitions at the borders of regions that might be involved in bacteriophage integration.
The most prominent duplication was a 190-bp element that was repeated 10 times in the chromosome. Multiple alignment of the ten repeats showed that they could be classified into two subfamilies with six and three copies each, plus a copy of what appears to be a chimaera. Similar sequences have also been described in the closely related species Bacillus licheniformis21,22. A striking feature of these repeats is that they are only found in half of the chromosome, at either side of the origin of replication, with five repeats on each side. Furthermore, with the exception of the most distal repeat at position 737,062, they lie in the same orientation with respect to the movement of the replication fork (Figs 2 (PDF File: 1,684k) and 3). Putative secondary structures conserved by compensatory mutations, as well as an insert in three of the copies, suggest that this element could indicate a structural RNA molecule.

The movement of the replication forks is represented by arrows. Ribosomal RNA operons are indicated by brown boxes. Known prophages and prophage-like elements are represented as blue lines. The 190-bp element repeated ten times is represented by green lines.
Analysis at the transcription and translation level. Over 4,000 putative protein coding sequences (CDSs) have been identified, with an average size of 890 bp, covering 87% of the genome sequence (Fig. 2 (PDF File: 1,684k)). We found that 78% of the genes started with ATG, 13% with TTG and 9% with GTG, which compares with 85%, 3% and 14%, respectively, in E. coli8. Fifteen genes (eight in the predicted CDSs in bacteriophage SPβ) exhibiting unusual start codons (namely ATT and CTG) were also identified through their similarities to known genes in other organisms or because they had a good GeneMark prediction (see Methods). This has not yet been substantiated experimentally. However, in the case of the gene coding for translation initiation factor 3, the similarity with its E. coli counterpart strongly suggests that the initiation codon is ATT, as is the case in E. coli.
We have not annotated CDSs that largely or entirely overlap existing genes, although such genes (for example, comS inside srfAA) certainly exist. It is also likely that some of the short CDSs present in the B. subtilis genome have been overlooked. For these reasons and possible sequencing errors, the estimated number of B. subtilis CDSs will fluctuate around the present figure of 4,100.
In several cases, in-frame termination codons or frameshifts were confirmed to be present on the chromosome (for example, an internal termination codon in ywtF, or the known programmed translational frameshift in prfB), indicating that the genes are either non-functional (pseudogenes) or subject to regulatory processes. It will therefore be of interest to determine whether these gene features are conserved in related Bacillus species, especially as strain 168 is derived from the Marburg strain that was subjected to X-ray irradiation23.
A few regions do not have any identifiable feature indicating that they are transcribed: they could be ‘grey holes’ of the type described in E. coli24. Preliminary studies involving all regions of more than 400 bp without annotated CDSs indicated that, of ∼300 such regions, only 15% were likely to be really devoid of protein-coding sequences. One of the longest such regions, located between yfjO and yfjN, is 1,628 bp long. Grey holes seem generally to be clustered near the terminus of replication. However, a grey-hole cluster located at ∼600 kb might be related to the temporary chromosome partition observed during the first stages of sporulation, when a segment of about one-third of the chromosome enters the prespore, and remains the sole part of the chromosome in the prespore for a significant transition period25.
The codon usage of B. subtilis CDSs was analysed using factorial correspondence analysis17. We found that the CDSs of B. subtilis could be separated into three well-defined classes (Fig. 4). Class 1 comprises the majority of the B. subtilis genes (3,375 CDSs), including most of the genes involved in sporulation. Class 2 (188 CDSs) includes genes that are highly expressed under exponential growth conditions, such as genes encoding the transcription and translation machineries, core intermediary metabolism, stress proteins, and one-third of genes of unknown function. Class 3 (537 CDSs) contains a very high proportion of genes of unidentified function (84%), and the members of this class have codons enriched in A+ T residues. These genes are usually clustered into groups between 15 and 160 genes (for example, bacteriophage SPβ) and correspond to the A+ T-rich islands described above (Fig. 1). When they are of known function, or when their products display similarity to proteins of known function, they usually correspond to functions found in, or associated with, bacteriophages or transposons, as well as functions related to the cell envelope. This includes the region ydc/ydd/yde (40 genes that are missing in some B. subtilis strains26), where gene products showing similarities to bacteriophage and transposon proteins are intertwined. Many of these genes are associated with virulence genes identified in pathogenic Gram-positive bacteria, suggesting that such virulence factors are transmitted horizontally among bacteria at a much higher frequency than previously thought. If we include these A+ T-rich regions as possible cryptic phages, together with known bacteriophages or bacteriophage-like elements (SPβ, PBSX and the skin element), we find that the genome of B. subtilis 168 contains at least 10 such elements (Figs 2 (PDF File: 1,684k) and 3). Annotation of the corresponding regions often reveals the presence of genes that are similar to bacteriophage lytic enzymes, perhaps accounting for the observation that B. subtilis cultures are extremely prone to lysis.

Red dots, genes from class 1; green triangles, genes from class 2; blue crosses, genes from class 3. Class 2 contains genes coding for the translation and transcription machineries, and genes of the core intermediary metabolism. Class 3 genes correspond to codons strongly enriched in A or T in the wobble position; they generally belong to prophage-like inserts in the genome.
The ribosomal RNA genes have been previously identified and shown to be organized into ten rRNA operons, mainly clustered around the origin of replication of the chromosome (Figs 2 (PDF File: 1,684k) and 3). In addition to the 84 previously identified tRNA genes, by using the Palingol27 and tRNAscan28 programs, we propose four putative new tRNA loci (at 1,262 kb, 1,945 kb, 2,003 kb and 2,899 kb), specific for lysine, proline and arginine (UUU, GGG, CCU and UCU anticodons, respectively). The 10S RNA involved in degradation of proteins made from truncated mRNA has been identified (ssrA), as well as the RNA component of RNase P (rnpB) and the 4.5S RNA involved in the secretion apparatus (scr).
There is a strong transcription orientation bias with respect to the movement of the replication fork: 75% of the predicted genes are transcribed in the direction of replication. Plotting the density of coding nucleotides in each strand along the chromosome readily identifies the replication origin and terminus (Fig. 3). To identify putative operons, we followed ref. 29 for describing Rho-independent transcription termination sites. This yielded ∼1,630 putative terminators (340 of which were bidirectional). We retained only those that were located less than 100 bp downstream of a gene, or that were considered by the program to be ‘very strong’ (in order to account for possible erroneous CDSs). This yielded a total of ∼1,250 terminators, with a mean operon size of three genes. A similar approach to the identification of promoters is problematical, especially because at least 14 sigma factors, recognizing different promoter sequences, have been identified in B. subtilis. Nevertheless, the consensus of the main vegetative sigma factor (σA) appears to be identical to its counterpart in E. coli (σ70): 5′-TTGACA- n17-TATAAT-3′. Relaxing the constraints of the similarity to sigma-specific consensus sequences led to an extremely high number of false-positive results, suggesting that the consensus-oriented approach to the identification of promoters should be replaced by another approach17.
Classification of gene products
Genes were classified according to ref. 14, based on the representation of cells as Turing machines in which one distinguishes between the machine and the program (Table 1 (PDF File: 275k)). Using the BLAST2P software running against a composite protein databank compound of SWISS-PROT (release 34), TREMBL (release 3, update 1) and B. subtilis proteins, we assigned at least one significant counterpart with a known function to 58% of the B. subtilis proteins. Thus for up to 42% of the gene products, the function cannot be predicted by similarity to proteins of known function: 4% of the proteins are similar only to other unknown proteins of B. subtilis; 12% are similar to unknown proteins from some other organism; and 26% of the proteins are not significantly similar to any other proteins in databanks. This preliminary analysis should be interpreted with caution, because only ∼1,200 gene functions (30%) have been experimentally identified in B. subtilis. We used the ‘y’ prefix in gene names to emphasize that the function has not been ascertained (2,853 ‘y’ genes, representing 70%).
Regulatory systems. Transcription regulatory proteins. Helix–turn–helix proteins form a large family of regulatory proteins found in both prokaryotes and eukaryotes. There are several classes, including repressors, activators and sigma factors. Using BLAST searches, we constructed consensus matrices for helix–turn–helix proteins to analyse the B. subtilis protein library. We identified 18 sigma or sigma-like factors, of which nine (including a new one) are of the SigA type. We also putatively identified 20 regulators (among which 18 were products of ‘y’ genes) of the GntR family, 19 regulators (15 ‘y’ genes) of the LysR family, and 12 regulators (5 ‘y’ genes) of the LacI family. Other transcription regulatory proteins were of the AraC family (11 members, 10 ‘y’), the Lrp family (7 members, 3 ‘y’), the DeoR family (6 members, 3 ‘y’), or additional families (such as the MarR, ArsR or TetR families). A puzzling observation is that several regulatory proteins display significant similarity to aminotransferases (seven such enzymes have been identified as showing similarity to repressors).
Two-component signal-transduction pathways.Two-component regulatory systems, consisting of a sensor protein kinase and a response regulator, are widespread among prokaryotes. We have identified 34 genes encoding response regulators in B. subtilis, most of which have adjacent genes encoding histidine kinases. Response regulators possess a well-conserved N-terminal phospho-acceptor domain30, whereas their C-terminal DNA-binding domains share similarities with previously identified response regulators in E. coli, Rhizobium meliloti, Klebsiella pneumoniae or Staphylococcus aureus. Representatives of the four subfamilies recently identified in E. coli31 (OmpR, FixJ, CitB and LytR) have been identified in B. subtilis. In a fifth subfamily, CheY, the DNA-binding domain is absent. The DNA-binding domain of a single B. subtilis response regulator, YesN, shares similarity with regulatory proteins of the AraC family.
Quorum sensing. The B. subtilis genome contains 11 aspartate phosphatase genes, whose products are involved in dephosphorylation of response regulators, that do not seem to have counterparts in Gram-negative bacteria such as E. coli. Downstream from the corresponding genes are some small genes, called phr, encoding regulatory peptides that may serve as quorum sensors32. Seven phr genes have been identified so far, including three new genes (phrG, phrI and phrK).
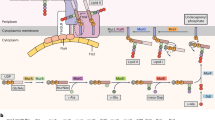
Protein secretion. It is known that B. subtilis and related Bacillus species, in particular B. licheniformis and B. amyloliquefaciens, have a high capacity to secrete proteins into the culture medium. Several genes encoding proteins of the major secretion pathway have been identified: secA, secD, secE, secF, secY, ffh and ftsY. Surprisingly, there is no gene for the SecB chaperone. It is thought that other chaperone(s) and targeting factor(s), such as Ffh and FtsY, may take over the SecB function. Further, although there is only one such gene in E. coli, five type I signal peptidase genes (sipS, sipT, sipU, sipV and sipW) have been found33. The lsp gene, encoding a type II signal peptidase required for processing of lipo-modified precursors, was also identified. PrsA, located at the outer side of the membrane, is important for the refolding of several mature proteins after their translocation through the membrane.
Other families of proteins.ABC transporters were the most frequent class of proteins found in B. subtilis. They must be extremely important in Gram-positive bacteria, because they have an envelope comprising a single membrane. ABC transporters will therefore allow such bacteria to escape the toxic action of many compounds. We propose that 77 such transporters are encoded in the genome. In general they involve the interaction of at least three gene products, specified by genes organized into an operon. Other families comprised 47 transport proteins similar to facilitators (and perhaps sometimes part of the ABC transport systems), 18 amino-acid permeases (probably antiporters), and at least 16 sugar transporters belonging to the PEP-dependent phosphotransferase system.
General stress proteins are important for the survival of bacteria under a variety of environmental conditions. We identified 43 temperature-shock and general stress proteins displaying strong similarity to E. coli counterparts.
Missing genes. Histone-like proteins such as HU and H-NS have been identified in E. coli. We found that B. subtilis encodes two putative histone-like proteins that show similarity to E. coli HU, namely HBsu and YonN, but found no homologue to H-NS. It is known that the hbs gene encoding HBsu is essential, but we do not expect the yonN gene to be essential because it is present in the SPβ prophage. IHF is similar to HU, and it is not known whether HBsu plays a similar role to that of IHF in E. coli. Similarly, no protein similar to FIS could be found.
Genes encoding products that interact with methylated DNA, such as seqA in E. coli, involved in the regulation of replication initiation timing, or mutH, the endonuclease recognizing the newly synthesized strand during mismatch repair at hemi-methylated GATC sites, are also missing. This is in line with the absence of known methylation in B. subtilis, equivalent to Dam methylation in E. coli. Similarly, E. coli sfiA, encoding an inhibitor of FtsZ action in the SOS response, has no counterpart in B. subtilis. In contrast, B. subtilis replication initiation-specific genes, such as dnaB and dnaD, are missing in E. coli. The exact counterpart of the E. coli mukB gene, involved in chromosome partitioning, does not exist in B. subtilis, but genes spo0J and smc (Smc is weakly similar to MukB), which are suggested to be involved in partitioning of the B. subtilis chromosome, are missing in E. coli.
Turnover of mRNA is controlled in E. coli by a ‘degradosome’ comprising RNase E. It has a counterpart in B. subtilis, but we failed to find a clear homologue of RNase E in this organism. Whether this is related to the role of ribosomal protein S1 as an RNA helicase involved in mRNA turnover in E. coli requires further investigation. In particular, a homologue of rpsA (S1 structural gene), ypfD, might be involved in a structure homologous to the degradosome34.
Structurally unrelated genes of similar function. Several genes encode products that have similar functions in E. coli and B. subtilis, but have no evident common structure. This is the case for the helicase loader genes, E. coli dnaC and B. subtilis dnaI; the genes coding for the replication termination protein, E. coli tus and B. subtilis rtp; and the division topology specifier genes, E. coli minE and B. subtilis divIVA. The situation may even be more complex in multisubunit enzymes: B. subtilis synthesizes two DNA polymerase III α chains, one having 3′–5′ proofreading exonuclease activity (PolC) and the other without the exonuclease activity (DnaE); in E. coli, only the latter exists. E. coli DNA polymerase II is structurally related to DNA polymerase α of eukaryotes, whereas B. subtilis YshC is related to DNA polymerase β.
Metabolism of small molecules
The type and range of metabolism used for the interconversion of low-molecular-weight compounds provide important clues to an organism's natural environment(s) and its biologil activity. Here we briefly outline the main metabolic pathways of B. subtilis before the reconstruction of these pathways in silico, the correlation of genes with specific steps in the pathway, and ultimately the prediction of patterns of gene expression.
Intermediary metabolism.It has long been known that B. subtilis can use a variety of carbohydrates. As expected, it encodes an Embden–Meyerhof–Parnas glycolytic pathway, coupled to a functional tricarboxylic acid cycle. Further, B. subtilis is also able to grow anaerobically in the presence of nitrate as an electron acceptor. This metabolism is, at least in part, regulated by the FNR protein, binding to sites upstream of at least eight genes (four sites experimentally confirmed and four putative sites). A noteworthy feature of B. subtilis metabolism is an apparent requirement of branched short-chain carboxylic acids for lipid biosynthesis35. Branched-chain 2-keto acid decarboxylase activity exists and may be linked to a variety of genes, suggesting that B. subtilis can synthesize and utilize linear branched short-chain carboxylic acids and alcohols.
Amino-acid and nucleotide metabolism. Pyrimidine metabolism of B. subtilis seems to be regulated in a way fundamentally different from that of E. coli, as it has two carbamylphosphate synthetases (one specific for arginine synthesis, the other for pyrimidine). Additionally, the aspartate transcarbamylase of B. subtilis does not act as an allosteric regulator as it does in E. coli. As in other microorganisms, pyrimidine deoxyribonucleotides are synthesized from ribonucleoside diphosphates, not triphosphates. The cytidine diphosphate required for DNA synthesis is derived from either the salvage pathway of mRNA turnover or from the synthesis of phospholipids and components of the cell wall. This means that polynucleotide phosphorylase is of fundamental importance in nucleic acid metabolism, and may account for its important role in competence36. Two ribonucleoside reductases, both of class I, NrdEF type, are encoded by the B. subtilis chromosome, in one case from within the SPβ genome. In this latter case, the gene corresponding to the large subunit both contains an intron and codes for an intein (V.L., unpublished data). The gene of the small subunit of this enzyme also contains an intron, encoding an endonuclease, as was found for the homologue in bacteriophage T4.
By similarity with genes from other organisms, there appears to be, in addition to genes involved in amino-acid degradation (such as the roc operon, which degrades arginine and related amino acids), a large number of genes involved in the degradation of molecules such as opines and related molecules, derived from plants. This is also in line with the fact that B. subtilis degrades polygalacturonate, and suggests that, in its biotope, it forms specific relations with plants.
Secondary metabolism. In addition to many genes coding for degradative enzymes, almost 4% of the B. subtilis genome codes for large multifunctional enzymes (for example, the srf, pps and pks loci), similar to those involved in the synthesis of antibiotics in other genera of Gram-positive bacteria such as Streptomyces. Natural isolates of B. subtilis produce compounds with antibiotic activity, such as surfactin, fengycin and difficidin, that can be related to the above-mentioned loci. This bacterium therefore provides a simple and genetically amenable model in which to study the synthesis of antibiotics and its regulation. These pathways are often organized in very long operons (for example, the pks region spans 78.5 kb, about 2% of the genome). The corresponding sequences are mostly located near the terminus of replication, together with prophages and prophage-like sequences.
Paralogues and orthologues
It is important to relate intermediary metabolism to genome structure, function and evolution. We therefore compared the B. subtilis proteins with themselves, as well as with proteins from known complete genomes, using a consistent statistical method that allows the evaluation of unbiased probabilities of similarities between proteins37,38. For Z-scores higher than 13, the number of proteins similar to each given protein does not vary, indicating that this cut-off value identifies sets of proteins that are significantly similar.
Families of paralogues. Many of the paralogues constitute large families of functionally related proteins, involved in the transport of compounds into and out of the cell, or involved in transcription regulation. Another part of the genome consists of gene doublets (568 genes), triplets (273 genes), quadruplets (168 genes) and quintuplets (100 genes). Finally, about half of the genome is made of genes coding for proteins with no apparent paralogues (Fig. 5). No large family comprises only proteins without any similarity to proteins of known function.

Each B. subtilis protein has been compared with all other proteins in the genome, using a Smith and Waterman algorithm. The baseline is established by making a similar comparison using 100 independent random shuffles of the protein sequence (Z-score > 13).
The process by which paralogues are generated is not well understood, but we might find clues by studying some of the duplications in the genome. Several approximate DNA repetitions, associated with very high levels of protein identity, were found, mainly within regions putatively or previously identified as prophages. This is in line with previous observations about PBSX and the skin element39,40, and suggests that these prophage-like elements share a common ancestor and have diverged relatively recently. In addition, several protein duplications are in genes that are located very close to each other, such as yukL and dhbF (the corresponding proteins are 65% identical in an overlap of 580 amino acids), yugJ and yugK (proteins 73% identical), yxjG and yxjH (proteins 70% identical), and the entire opuB operon, which is duplicated 3 kb away (opuC operon, yielding ∼80% of amino-acid identity in the corresponding proteins).
The study of paralogues showed that, as in other genomes, a few classes of genes have been highly expanded. This argues against the idea of the genome evolving through a series of duplications of ancestral genomes, but rather for the idea of genes as living organisms, subject to evolutionary constraints, some being submitted to expansion and natural selection, and others to local duplications of DNA regions.
Among paralogue doublets, some were unexpected, such as the three aminoacyl tRNA synthetases doublets (hisS (2,817 kb) and hisZ (3,588 kb); thrS (2,960 kb) and thrZ (3,855 kb); tyrS (3,036 kb) and tyrZ (3,945 kb)) or the two mutS paralogues (mutS and yshD). This latter situation is similar to that found in Synechocystis. In the case of B. subtilis, the presence of two MutS proteins could indicate that there are two different pathways for long-patch mismatch repair, possibly a consequence of the active genetic transformation mechanism of B. subtilis.
Families of orthologues. Because Mycoplasma spp. are thought to be derived from Gram-positive bacteria similar to B. subtilis, we compared the B. subtilis genome with that of M. genitalium. Among the 450 genes encoded by M. genitalium, the products of 300 are similar to proteins of B. subtilis. Among the 146 remaining gene products, a further 3 are similar to proteins of other Bacillus species, and 9 to proteins of other Gram-positive bacteria; 25 are similar to proteins of Gram-negative bacteria; and 19 are similar to proteins of other Mycoplasma spp. This leaves only 90 genes that would be specific to M. genitalium and might be involved in the interaction of this organism with its host.
The B. subtilis genome is similar in size to that of E. coli. Because these bacteria probably diverged more than one billion years ago, it is of evolutionary value to investigate their relative similarity. About 1,000 B. subtilis genes have clear orthologous counterparts in E. coli (one-quarter of the genome). These genes did not belong either to the prophage-like regions or to regions coding for secondary metabolism (∼15% of the B. subtilis genome). This indicates that a large fraction of these genomes shared similar functions. At first sight, however, it seems that little of the operon structure has been conserved. We nevertheless found that ∼100 putative operons or parts of operons were conserved between E. coli and B. subtilis. Among these, ∼12 exhibited a reshuffled gene order (typically, the arabinose operon is araABD in B. subtilis and araBAD in E. coli). In addition to the core of the translation and transcription machinery, we identified other classes of operons that were well conserved between the two organisms, including major integrated functions such as ATP synthesis (atp operon) and electron transfer (cta and qox operons). As well as being well preserved, the murein biosynthetic region was partly duplicated, allowing creation of part of the genes required for the sporulation division machinery41. The amino-acid biosynthesis genes differ more in their organization: the E. coli genes for arginine biosynthesis are spread throughout the chromosome, whereas the arginine biosynthesis genes of B. subtilis form an operon. The same is true for purine biosynthetic genes. Genes responsible for the biosynthesis of coenzymes and prosthetic groups in B. subtilis are often clustered in operons that differ from those found in E. coli. Finally, several operons conserved in E. coli and B. subtilis correspond to unknown functions, and should therefore be priority targets for the functional analysis of these model genomes.
Comparison with Synechocystis PCC6803 revealed about 800 orthologues. However, in this case the putative operon structure is extremely poorly conserved, apart from four of the ribosomal protein operons, the groES–groEL operon, yfnHG (respectively in Synechocystis rfbFG), rpsB-tsf, ylxS-nusA-infB, asd-dapGA-ymfA, spmAB, efp-accB, grpE-dnaK, yurXW. The nine-gene atp operon of B. subtilis is split into two parts in Synechocystis: atpBE and atpIHGFDAC.
Conclusion
The biochemistry, physiology and molecular biology of B. subtilis have been extensively studied over the past 40 years. In particular, B. subtilis has been used to study postexponential phase phenomena such as sporulation and competence for DNA uptake. The genome sequences of E. coli and B. subtilis provide a means of studying the evolutionary divergence, one billion years ago, of eubacteria into the Gram-positive and Gram-negative groups. The availability of powerful genetic tools will allow the B. subtilis genome sequence data to be exploited fully within the framework of a systematic functional analysis program, undertaken by a consortium of 19 European and 7 Japanese laboratories coordinated by S. D. Ehrlich (INRA, Jouy-en-Josas, France) and by N. Ogasawara and H. Yoshikawa (Nara Institute of Science and Technology, Nara, Japan).
Methods
Genome cloning and sequencing. An international consortium was established to sequence the genome of B. subtilis strain 168 (refs 9, 10, 42). At its peak, 25 European, seven Japanese and one Korean laboratory participated in the program, together with two biotechnology companies. Five contiguous DNA regions totalling 0.94 Mb, and two additional regions of 0.28 and 0.14 Mb, were sequenced by the Japanese partners, while the European partners sequenced a total of 2.68 Mb. A few sequences from strain 168 published previously were not resequenced when long overlaps did not indicate differences.
A major technical difficulty was the inability to construct in E. coli gene banks representative of the entire B. subtilis chromosome using vectors that have proved efficient for other sources of bacterial DNA (such as bacteriophage or cosmid vectors). This was due to the generally very high level of expression of B. subtilis genes in E. coli, leading to toxic effects. This limitation was overcome by: cloning into a variety of vectors9,43,44; using an E. coli strain maintaining low-copy number plasmids44; using an integrative plasmid/marker rescue genome-walking strategy44; and in vitro amplification using polymerase chain reaction (PCR) techniques45,46.
Although cloning vectors were used in the early stages as templates for sequencing reactions, they were largely superseded in the later stages by long-range and inverse PCR techniques. To reduce sequencing errors resulting from PCR amplification artefacts, at least eight amplification reactions were performed independently and subsequently pooled. The various sequencing groups were free to choose their own strategy, except that all DNA sequences had to be determined entirely on both strands.
Sequence annotation and verification. The sequences were annotated by the groups, and sent to a central depository at the Institut Pasteur14. The Japanese sequences were also sent there through the Japanese depository at the Nara Institute of Science and Technology. The same procedures were used to identify CDSs and to detect frameshifts. They were embedded within a cooperative computer environment dedicated to automatic sequence annotation and analysis39. In a first step, we identified in all six possible frames the open reading frames (ORFs) that were at least 100 codons in length. In a second step, three independent methods were used: the first method used the GeneMark coding-sequence prediction method47 together with the search for CDSs preceded by typical translation initiation signals (5′-AAGGAGGTG-3′), located 4–13 bases upstream of the putative start codons (ATG, TTG or GTG); the second method used the results of a BLAST2X analysis performed on the entire B. subtilis genome against the non-redundant protein databank at the NCBI; and the third method was based on the distribution of non-overlapping trinucleotides or hexanucleotides in the three frames of an ORF48.
In general, frameshifts and missense mutations generating termination codons or eliminating start codons are relatively easy to detect. We shall devise a procedure for detecting another type of error, GC instead of CG or vice versa, which are much more difficult to identify. It should be noted that putative frameshift errors should not be corrected automatically. The sequences of the flanking regions of a 500-bp fragment centred around a putative error were sent to an independent verification group, which performed PCR amplifications using chromosomal DNA as template, and sequenced the corresponding DNA products.
Organization and accessibility of data. The B. subtilis sequence data have been combined with data from other sources (biochemical, physiological and genetic) in a specialized database, SubtiList49, available as a Macintosh or Windows stand-alone application (4th Dimension runtime) by anonymous FTP at ftp://ftp.pasteur.fr/pub/GenomeDB/SubtiList. SubtiList is also accessible through a World-Wide Web server at http://www.pasteur.fr/Bio/SubtiList.html, where it has been implemented on a UNIX system using the Sybase relational database management system. A completely rewritten version of SubtiList is in preparation to facilitate browsing of the information of the whole chromosome. Flat files of the whole DNA and protein sequences in EMBL and FASTA format will be made available at the above ftp address. Another B. subtilis genome database is also under development at the Human Genome Center of Tokyo University (http://www.genome.ad.jp), and SubtiList will also be available there.

Arrows indicate the orientation of transcription. Genes are coloured according to their classification into six broad functional categories (blue, category I; green, category II; red, category III; orange, category IV; purple, category V; pink, category VI; see Table 1). Class 2 CDSs according to codon usage analysis are indicated by oblique hatches, and class 3 CDSs are indicated by vertical hatches. Ribosomal RNA genes are coloured in yellow. Transfer RNA genes are marked by triangles. Other RNA genes are represented as white arrows. Known genes (non-‘y’ genes) are printed in bold type. Putative transcription termination sites are represented as loops. Known prophages and prophage-like elements are indicated by brown hatches on the chromosome line. The 190-bp element repeated ten times is represented by hatched boxes.
References
Fleischmann, R. D. et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269, 496–512 (1995).
Fraser, C. M. et al. The minimal gene complement of Mycoplasma genitalium. Science 270, 397–403 (1995).
Kaneko, T. et al. Sequence analysis of the genome of the unicellular Cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 3, 109–136 (1996).
Bult, C. J. et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 273, 1058–1073 (1996).
Himmelreich, R. et al. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 24, 4420–4449 (1996).
Goffeau, A. et al. The yeast genome directory. Nature 387, 5–105 (1997).
Tomb, J.-F. et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547 (1997).
Blattner, F. R. et al. The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462 (1997).
Kunst, F., Vassarotti, A. & Danchin, A. Organization of the European Bacillus subtilis genome sequencing project. Microbiology 389, 84–87 (1995).
Ogasawara, N. & Yoshikawa, H. The systematic sequencing of the Bacillus subtilis genome in Japan. Microbiology 142, 2993–2994 (1996).
Harwood, C. R. Bacillus subtilis and its relatives: molecular biological and industrial workhorses. Trends Biotechnol. 10, 247–256 (1992).
Stragier, P. & Losick, R. Molecular genetics of sporulation in Bacillus subtilis. Annu. Rev. Genet. 30, 297–341 (1996).
Solomon, J. M. & Grossman, A. D. Who's competent and when: regulation of natural genetic competence in bacteria. Trends Genet. 12, 150–155 (1996).
Moszer, I., Kunst, F. & Danchin, A. The European Bacillus subtilis genome sequencing project: current status and accessibility of the data from a new World Wide Web site. Microbiology 142, 2987–2991 (1996).
Franks, A. H., Griffiths, A. A. & Wake, R. G. Identification and characterization of new DNA replication terminators in Bacillus subtilis. Mol. Microbiol. 17, 13–23 (1995).
Lobry, J. R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 13, 660–665 (1996).
Hénaut, A. & Danchin, A. in Escherichia coli and Salmonella: Cellular and Molecular Biology (eds Neidhardt, F. et al.) 2047–2066 (ASM, Washington DC, (1996)).
Nussinov, R. The universal dinucleotide asymmetry rules in DNA and amino acid codon choice. Nucleic Acids Res. 17, 237–244 (1981).
Karlin, S., Burge, C. & Campbell, A. M. Statistical analyses of counts and distributions of restriction sites in DNA sequences. Nucleic Acids Res. 20, 1363–1370 (1992).
Burge, C., Campbell, A. M. & Karlin, S. Over- and under-representation of short oligonucleotides in DNA sequences. Proc. Natl Acad. Sci. USA 89, 1358–1362 (1992).
Kasahara, Y., Nakai, S. & Ogasawara, H. Sequen analysis of the 36-kb region between gntZ and trnY genes of Bacillus subtilis genome. DNA Res. 4, 155–159 (1997).
Presecan, E. et al. The Bacillus subtilis genome from gerBC (311°) to licR (334°). Microbiology 143, 3313–3328 (1997).
Burkholder, P. R. & Giles, N. H. Induced biochemical mutations in Bacillus subtilis. Am. J. Bot. 33, 345–348 (1947).
Daniels, D. L., Plunkett, G. II, Burland, V. & Blattner, F. R. Analysis of the Escherichia coli genome: DNA sequence of the region from 84.5 to 86.5 minutes. Science 257, 771–778 (1992).
Wu, L. J. & Errington, J. Bacillus subtilis SpoIIIE protein required for DNA segregation during asymmetric cell division. Science 264, 572–575 (1994).
Itaya, M. Stability and asymmetric replication of the Bacillus subtilis 168 chromosome structure. J. Bacteriol. 175, 741–749 (1993).
Billoud, B., Kontic, M. & Viari, A. Palingol: a declarative programming language to describe nucleic acids' secondary structures and to scan sequence database. Nucleic Acids Res. 24, 1395–1403 (1996).
Fichant, G. A. & Burks, C. Identifying potential tRNA genes in genomic DNA sequences. J. Mol. Biol. 220, 659–671 (1991).
d'Aubenton Carafa, Y., Brody, E. & Thermes, C. Prediction of rho-independent Escherichia coli transcription terminators. A statistical analysis of their RNA stem-loop structures. J. Mol. Biol. 216, 835–858 (1990).
Stock, J. B., Surette, M. G., Levitt, M. & Park, P. in Two-Component Signal Transduction (eds Hoch, J. A. & Silhavy, T. J.) 25–51 (ASM, Washington DC, (1995)).
Mizuno, T. Compilation of all genes encoding two-component phosphotransfer signal transducers in the genome of Escherichia coli. DNA Res. 4, 161–168 (1997).
Perego, M., Glaser, P. & Hoch, J. A. Aspartyl-phosphate phosphatases deactivate the response regulator components of the sporulation signal transduction system in Bacillus subtilis. Mol. Microbiol. 19, 1151–1157 (1996).
Tjalsma, H. et al. Bacillus subtilis contains four closely related type I signal peptidases with overlapping substrate specificities: constitutive and temporally controlled expression of different sip genes. J. Biol. Chem. 272, 25983–25992 (1997).
Danchin, A. Comparison between the Escherichia coli and Bacillus subtilis genomes suggests that a major function of polynucleotide phosphorylase is to synthesize CDP. DNA Res. 4, 9–18 (1997).
Suutari, M. & Laakso, S. Unsaturated and branched chain-fatty acids in temperature adaptation of Bacillus subtilis and Bacillus megaterium. Biochim. Biophys. Acta 1126, 119–124 (1992).
Luttinger, A., Hahn, J. & Dubnau, D. Polynucleotide phosphorylase is necessary for competence development in Bacillus subtilis. Mol. Microbiol. 19, 343–356 (1996).
Landès, C., Hénaut, A. & Risler, J.-L. Acomparison of several similarity indices used in the classification of protein sequences: a multivariate analysis. Nucleic Acids Res. 20, 3631–3637 (1992).
Glémet, E. & Codani, J.-J. LASSAP, a LArge Scale Sequence compArison Package. Comput. Appl. Biosci. 13, 137–143 (1997).
Médigue, C., Moszer, I., Viari, A. & Danchin, A. Analysis of a Bacillus subtilis genome fragment using a co-operative computer system prototype. Gene 165, GC37–GC51 (1995).
Krogh, S., O'Reilly, M., Nolan, N. & Devine, K. M. The phage-like element PBSX and part of the skin element, which are resident at different locations on the Bacillus subtilis chromosome, are highly homologous. Microbiology 142, 2031–2040 (1996).
Daniel, R. A., Drake, S., Buchanan, C. E., Scholle, R. & Errington, J. The Bacillus subtilis spoVD gene encodes a mother-cell-specific penicillin-binding protein required for spore morphogenesis. J. Mol. Biol. 235, 209–220 (1994).
Anagnostopoulos, C. & Spizizen, J. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 81, 741–746 (1961).
Azevedo, V. et al. An ordered collection of Bacillus subtilis DNA segments cloned in yeast artificial chromosomes. Proc. Natl Acad. Sci. USA 90, 6047–6051 (1993).
Glaser, P. et al. Bacillus subtilis genome project: cloning and sequencing of the 97 kb region from 325° to 333°. Mol. Microbiol. 10, 371–384 (1993).
Ogasawara, N., Nakai, S. & Yoshikawa, H. Systematic sequencing of the 180 kilobase region of the Bacillus subtilis chromosome containing the replication origin. DNA Res. 1, 1–14 (1994).
Sorokin, A. et al. Anew approach using multiplex long accurate PCR and yeast artificial chromomes for bacterial chromosome mapping and sequencing. Genome Res. 6, 448–453 (1996).
Borodovsky, M. & McIninch, J. GENMARK: parallel gene recognition for both DNA strands. Comput. Chem. 17, 123–133 (1993).
Fichant, G. A. & Quentin, Y. Aframeshift error detection algorithm for DNA sequencing projects. Nucleic Acids Res. 23, 2900–2908 (1995).
Moszer, I., Glaser, P. & Danchin, A. SubtiList: a relational database for the Bacillus subtilis genome. Microbiology 141, 261–268 (1995).
Acknowledgements
We thank C. Anagnostopoulos, R. Dedonder and J. Hoch for their pioneering efforts, and A. Bairoch for advice in annotating B. subtilis protein data. The main funding of the European network was provided by the European Commission under the Biotechnology program. The Japanese project was included in the Human Genome Program, and supported by a research grant from the Ministry of Education, Science and Culture, and the Proposal-Based Advanced Industrial Technology R&D Program from New EEnergy and Industrial Technology Development Organization. The Swiss and Korean projects were funded by the Swiss National Fund and the Korean government, respectively. An industrial platform was set up to facilitate contacts between participants of the European consortium and some European biotechnology companies: DuPont de Nemours (France, USA), Frimond (Belgium), Genencor (Finland, USA), Gist Brocades (The Netherlands), Glaxo-Wellcome (UK, Italy), Hoechst Marion Roussel (France, Germany), F. Hoffmann-La Roche AG (Switzerland), Novo Nordisk (Denmark), SmithKline Beecham (UK).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Kunst, F., Ogasawara, N., Moszer, I. et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390, 249–256 (1997). https://doi.org/10.1038/36786
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/36786
This article is cited by
-
Construction of recombinant Lactococcus expressing thymosin and interferon fusion protein and its application as an immune adjuvant
Microbial Cell Factories (2024)
-
Research advances in the identification of regulatory mechanisms of surfactin production by Bacillus: a review
Microbial Cell Factories (2024)
-
Direct observation of a crescent-shape chromosome in expanded Bacillus subtilis cells
Nature Communications (2024)
-
Unveiling antibiofilm potential: proteins from Priestia sp. targeting Staphylococcus aureus biofilm formation
Antonie van Leeuwenhoek (2024)
-
Systematic analysis of drug combinations against Gram-positive bacteria
Nature Microbiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
