
Abstract
Campylobacter jejuni, from the delta-epsilon group of proteobacteria, is a microaerophilic, Gram-negative, flagellate, spiral bacterium—properties it shares with the related gastric pathogen Helicobacter pylori. It is the leading cause of bacterial food-borne diarrhoeal disease throughout the world1. In addition, infection with C. jejuni is the most frequent antecedent to a form of neuromuscular paralysis known as Guillain–Barré syndrome2. Here we report the genome sequence of C. jejuni NCTC11168. C. jejuni has a circular chromosome of 1,641,481 base pairs (30.6% G+C) which is predicted to encode 1,654 proteins and 54 stable RNA species. The genome is unusual in that there are virtually no insertion sequences or phage-associated sequences and very few repeat sequences. One of the most striking findings in the genome was the presence of hypervariable sequences. These short homopolymeric runs of nucleotides were commonly found in genes encoding the biosynthesis or modification of surface structures, or in closely linked genes of unknown function. The apparently high rate of variation of these homopolymeric tracts may be important in the survival strategy of C. jejuni.
Similar content being viewed by others
Main
Human infection is usually acquired by the consumption of contaminated food (especially poultry) or water1. Motile campylobacters colonize the intestines of a wide range of animals, but in immunologically naive humans infection frequently results in an inflammatory enterocolitis. The number of cases of Campylobacter infection reported in England and Wales in 1998 increased by 17% from the previous year, with the number of reported cases now more than double that due to Salmonella3. Despite its importance, effective control of Campylobacter in the food chain and the design of disease prevention strategies are hindered by a poor understanding of the genetics, physiology and virulence of this organism.
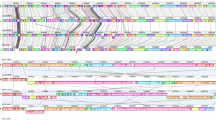
The genome of C. jejuni NCTC11168 is 1,641,481 base pairs (bp) in length. Of the 1,654 predicted coding sequences (CDS), at least 20 probably represent pseudogenes; the average gene length is 948 bp, and 94.3% of the genome codes for proteins, making it the densest bacterial genome sequenced to date. The bias towards G on the leading strand of the chromosome4 indicates that the origin of replication is near to the start of the dnaA gene. Strand bias is also evident in the CDSs; overall, 61.1% are transcribed in the same direction as replication (Fig. 1). We discovered two large regions of lower G+C content that encompass CDSs Cj1135–Cj1148 (25.4%) and Cj1421–Cj1442 (26.5%); these correspond to genes within the lipooligosaccharide (LOS) and extracellular polysaccharide (EP) biosynthesis clusters, respectively. Functional information (matches to genes of known function, or informative hydrophobicity profiles) could be deduced for 77.8% of the 1,654 CDSs, wheras 13.5% matched genes of unknown function in the database and 8.7% had no database match, or other functional information. The unusually low number of unknowns reflects the preponderance of predicted membrane, periplasmic and lipoproteins; these make up 10.3%, 7.8% and 2.3 % of the CDSs, respectively, and many of these have no database matches.

From the outside to the inside, the first circle shows coding sequences transcribed in the clockwise direction in dark green; the second shows coding sequences in the anticlockwise direction in pale green. The putative origin of replication is marked. The third shows the positions of hypervariable sequences in black, and the fourth and fifth show genes involved in the production of surface structures: clockwise in dark red and anticlockwise in pale red. The innermost histogram shows the similarity of each gene to its H. pylori orthologue, where present; the height of the bar, and the intensity of the colour, are proportional to the degree of similarity. The clusters of genes responsible for LOS biosynthesis, EP biosynthesis and flagellar modification are marked.
One surprising feature of the C. jejuni genome is the almost complete lack of repetitive DNA sequences. In fact, there are only four repeated sequences within the entire genome; three copies of the ribosomal RNA operon (6 kilobases (kb)) and three duplicated or triplicated CDSs. Apart from Cj0752, which is similar to part of IS605 tnpB from H. pylori, there is no evidence of any functional inserted sequence (IS) elements, transposons, retrons or prophages in the genome. Another intriguing feature of the genome is that, apart from salient exceptions such as the two ribosomal protein operons and gene clusters involved in LOS biosynthesis, EP biosynthesis and flagellar modification, there appears to be little organization of genes into operons or clusters. Although genes do fall into long, apparently linked sets, generally the genes within these sets appear to be functionally unrelated. The distribution of the genes involved in amino-acid biosynthesis, for example, reflects this organization: some of the his, leu and trp genes are apparently organized into operons, but the aro, asp, dap, gln, gly, ilv, met, phe, pro, ser, thr and tyr genes are scattered randomly throughout the genome.
The shotgun assembly revealed regions in which the sequences of otherwise identical clones varied at a single point. These were mainly, but not exclusively, length variations in polyG:C tracts (Table 1). The degree of variation differs between sites, and there are homopolymeric tracts that do not display variability within the observed shotgun sequences (Table 2). Variation in the length of polyG:C tracts is frequently associated with contingency genes in other pathogenic bacteria, and may be produced by slipped-strand mispairing during replication5; it can affect translation and has been shown to be responsible for phase variation of surface properties or antigenicity6. The appearance of variants due to slipped-strand mispairing occurs in Neisseria menigitidis with a frequency of 10-3 per cell per generation7; these variants are therefore likely to be rare in the sampled clones of a 10-fold shotgun library derived from a clonal population. The C. jejuni sequence, by contrast, shows some regions where three or more variants are present in almost equal proportions, in addition to many regions where two variants are present. The absolute rate of variation is difficult to estimate from these data, although the number of changes seen suggests that the frequency is much higher than in other organisms.
The variation in homopolymeric tracts was not an artefact of the sequencing process, as fourfold resequencing of several variants did not show any difference from the original sequence. A shotgun sequence of an appropriate lambda clone demonstrated that the variation did not occur during subcloning in Escherichia coli. Support for the existence of rapid phase variation in C. jejuni is also provided by H. pylori J99, for which similar variation was seen8. This rapid sequence variation suggests that C. jejuni may be lacking in DNA repair functions, indeed, many DNA repair genes studied in E. coli cannot be found in C. jejuni, including the direct repair genes ada and phr; the glycosylases tag, alkA, mutM and nfo; the mismatch repair genes vsr, mutH, mutL and, sbcB; and the SOS response genes lexA, umuC and umuD. Significantly, transcription-coupled repair may influence the rate of phase variation in N. meningitidis7. The high levels of variation seen in the shotgun sequences mean that it is not possible to produce a single definitive sequence for the C. jejuni genome. As such, it possesses some of the properties of a quasispecies; a phenomenon that is well described in RNA viruses9.
Most of the hypervariable sequences cluster on the genome and are coincident with the clusters of genes responsible for LOS biosynthesis, EP biosynthesis and flagellar modification (Fig. 1). Some of the variable genes can be ascribed putative functions, such as glycosyl-transferases; several others belong to two families (designated as 617 and 1318; Fig. 2, Table 3). The 617 family of genes have no homologues outside C. jejuni, while the 1318 family has two homologues in H. pylori (HP0114 and HP0465). Sixteen additional members of the 1318 family occur in various bacterial and archaeal species, none with a well-defined function; however, family members within the enterobacteriaceae are found within lipopolysaccharide gene clusters, supporting our hypothesis that these proteins are involved in the synthesis of surface structures. The rapid variation in surface properties implied by these results may have more relevance to the colonization of a dynamic intestinal environment than to immune avoidance.

Amino acids encoded by the hypervariable homopolymeric tracts are in boxes. Cj0617 and Cj0618, and Cj1335 and Cj1336 represent coding sequences frameshifted at the homopolymeric tract.
C. jejuni has been reported to produce a variety of toxins whose activity and/or role in pathogenesis remain controversial10. The genome of NCTC11168 does not contain a cholera-like toxin gene, although genes encoding the cytolethal distending toxin (cdtA-C) are present. A member of the family of contact-dependent haemolysins found in pathogenic Serpulina and Mycobacterium species11 (Cj0588), a putative integral membrane protein with a haemolysin domain (Cj0183) and a phospholipase (pldA) were also identified.
In contrast to most lipid A and some inner core biosynthesis genes, many LOS biosynthesis proteins are encoded by a large gene cluster (Cj1119–Cj1152) which has a role in core biosynthesis and also protein glycosylation12,13. There is increasing evidence that C. jejuni synthesizes an EP that is not attached to lipid A13, and thus the presence of genes similar to those involved in bacterial capsule biogenesis is significant. Two groups containing kps orthologues (involved in transport; A.V.K., manuscript submitted) were found and the region between the groups contains many different polysaccharide biosynthetic genes, some required for the biosynthesis of bacterial capsules. As might be expected with the presence of kps genes, no orthologues to wzx, wzy and wzz genes, which are essential for heteropolymeric O-chain biosynthesis, are present. Unusually, C. jejuni has three sets of neu genes involved in sialic acid biosynthesis. Sialic acid is an uncommon constituent of bacterial surface structures and, through molecular mimicry, may be important for evasion of host immunity and in post-infection autoimmune diseases such as Guillain–Barré syndrome. A complete cluster (neuB1, C1, A1) has a role in LOS sialylation (D. Linton et al., personal communication). Another set (neuB2 and C2) is in close proximity to ptmAB and may be involved with these genes in post-translational modification of flagellin14.
There are no obvious orthologues of the extensive Hop porin family of H. pylori15 and, contrary to a recent report16, no type III secretion systems were identified other than the flagellin export apparatus. In the absence of such a system, it is possible that the CiaB protein16 is secreted by the flagellin export apparatus17. In contrast to Campylobacter fetus and Campylobacter rectus, genes encoding S-layer proteins were apparently absent in C. jejuni. Under certain conditions, C. jejuni produce 4–7 nm wide filaments resembling pili18; although structural pilin orthologues appear to be absent, there are several type 4 pilus related genes such as a pre-pilin peptidase (Cj0825) and several putative type II export genes (Cj1470c–Cj1474c). Flagella are one of the best-defined virulence factors, as flaA and flaB mutants are markedly reduced in virulence19. Whether this is due to a direct involvement of the flagellin, or perhaps an inability to secrete proteins through the flagella export apparatus, is unknown17. Two further structural flagellin paralogues were identified (flaC Cj0720c and flaD Cj0887c), and orthologues of the majority of flagellar-associated genes of clearly understood function in bacteria such as S. typhimurium are represented; however, several genes involved in regulation are absent, including flhCD, flgM, fliT and fliK. These differences from the enterobacterial paradigm mirror those found in H. pylori. However, the gene encoding the H. pylori flagellar sheath protein hpaA (ref. 20) is absent in C. jejuni, as is a flagellar sheath.
Chemotaxis is important for intestinal colonization by C. jejuni. Like H. pylori, C. jejuni produces three proteins containing the response regulatory domain of CheY. One is CheY (Cj1118)21, and the others are found fused to a histidine protein kinase domain (CheA) or a CheW-like domain (CheV). In contrast to H. pylori, only one CheV orthologue is present. The regulation of CheY activity in C. jejuni is different from that of H. pylori as, unlike H. pylori, orthologues of both CheB and CheR are present. No orthologues of CheZ or other chemotaxis genes were found. Ten genes contain methyl-accepting chemotaxis protein domains; these are candidate chemoreceptor genes, some of which may transduce signals to non-taxis-associated pathways; only three (Cj1506, Cj0448, Cj0019) have orthologues in H. pylori.
The genome encodes five major iron-acquisition systems, which are mostly organized in operons under the control of the Fur protein22. Two of these iron-acquisition systems, the enterochelin uptake operon ceuBCDE and the siderophore receptor orthologue cfrA, have been described previously. In addition, there is a predicted haemin uptake operon chu (Cj1614–1617)22, a periplasmic binding protein dependent system (Cj0173c–0175c) and a siderophore receptor (Cj0178) with accessory genes. Three copies of the accessory tonB, exbB and exbD genes, which form the energy transduction machinery for transport of iron compounds over the outer membrane, were found. One of the exbB-exbD-tonB triplets follows the Cj0178 siderophore receptor, whereas another tonB gene is divergently orientated to cfrA. C. jejuni might therefore use separate energy transduction mechanisms for transport of the different iron substrates, as has been shown for Vibrio cholerae.
C. jejuni appears to have a broader repertoire of regulatory systems than H. pylori, which has a similar sized genome. Given that C. jejuni is found in a more diverse range of ecological niches than H. pylori, this might be expected. The apparent lack of operon organization raises fundamental issues concerning transcriptional regulation in Campylobacter . Although it is possible that each gene is independently transcribed, strand-specific gene grouping suggests co-transcription. Like H. pylori , the Campylobacter genome contains only three predicted sigma factors (rpoD, rpoN and fliA). The largest proportion of regulatory genes consists of members of the two-component regulator family. As in Bacillus subtilis, C. jejuni has an additional member of the Fur22 family, PerR, which regulates the peroxide stress regulon23. Unlike H. pylori, C. jejuni contains a possible crp/fnr family member (Cj0466) with both helix–turn–helix and cNMP-binding motifs.
As might be expected from the inability of C. jejuni to use carbohydrates as carbon or energy sources, very few genes for degradation of carbohydrates or amino acids were detected. The glycolytic pathway is also apparently incomplete; orthologues of the glucokinase and 6-phosphofructokinase genes were not found, although these functions may well be supplied by non-orthologous genes. Despite this, C. jejuni does appear to have all the genes necessary for gluconeogenesis and, unlike H. pylori, C. jejuni appears to encode an intact tricarboxylic acid cycle (like H. pylori, C. jejuni uses 2-oxoglutarate ferredoxin oxidoreductase (OorDABC)24, rather than 2-oxoglutarate dehydrogenase to interconvert 2-oxoglutarate and succinyl CoA).
C. jejuni and H. pylori are closely related by 16S rRNA phylogeny, share many biological properties and were previously classified within the Campylobacter genus. Figure 1 (and Supplementary Information) indicates which C. jejuni genes are present or absent in H. pylori 26695 (ref. 15). The three polysaccharide biosynthetic loci relating to surface structure stand out as being unique to C. jejuni and are correlated with a high density of polymorphic sequences. C. jejuni also contains a large number of biosynthetic genes not present in H. pylori, such as those directing the synthesis of purines, thiamine and many amino acids. Genes present in H. pylori but absent in C. jejuni include the urease operon, nickel transport system, vacuolating toxin and the Cag pathogenicity island15. These features are consistent with the unique niche of H. pylori in the stomach, and its pathophysiology and propensity for chronic infection.
Despite the close phylogenetic relationship of C. jejuni and H. pylori, strong similarities between them are mainly confined to housekeeping functions; only 55.4% of C. jejuni genes have orthologues in H. pylori. In most functions related to survival, transmission and pathogenesis, the organisms have remarkably little in common. This indicates that selective pressures have driven profound evolutionary changes to create two very different and specific pathogens appropriate to their niches, from a relatively close common ancestor. Overall, 28.0% of genes show closest similarity to genes from E. coli, 27.0% to genes from B. subtilis, 4.6% to genes from Archeoglobus fulgidus and 2.1% to genes from Saccharomyces cerevisiae. Several genes were found which have homologues only in the eukaryotic domain; these include the dUTPase (dut) gene (Cj1451), which is similar to those of Leishmania major and Trypanosoma cruzi only; there is no orthologue of the E. coli dut gene. Taken together, these statistics suggest that the evolution of the C. jejuni genome does not neatly mirror that of its small subunit ribosomal RNA, and that the placement of C. jejuni within the Gram-negative proteobacteria may rely on a simplified view of the evolutionary origin of its genome.
This genomic sequence provides the resources for a complete and detailed analysis of the pathogenic potential of this enigmatic pathogen. New insights into the biology of C. jejuni include the identification of hypervariable sequences, lack of classical operon structure and repetitive DNA, and an unexpected capacity for polysaccharide production.
Methods
A single colony of C. jejuni (NCTC11168, human origin, serotype O2, minimally passaged) was spread on one petri dish of CCDH agar (Campylobacter selective agar, Oxoid), and re-streaked on 10 CCDH agar plates. Cells were harvested and total DNA (10 mg) was isolated using proteinase K treatment followed by a phenol extraction procedure. The DNA was fragmented by sonication, size-fractionated on an agarose gel, and seven libraries were generated in pUC18 using size fractions ranging from 1.0 kb to 2.2 kb. Roughly 19,400 pUC clones were sequenced from both ends, using Dye-terminator chemistry on ABI 373 and 377 sequencing machines. The final assembly was generated from 33,900 reads, giving an ∼10-fold coverage of the genome. Sequence assembly was accomplished using Phrap (P. Green, unpublished), and the sequencing was finished using GAP425. The assembly was verified by genomic PCR reactions across all repeats, in addition to 130 forward and reverse reads from a random library of ∼10–16-kb fragments of genomic DNA cloned in lambdaFixII (Stratagene). In the final assembly, 0.13% of the genome was covered by a single clone only, and 0.11% was not sequenced on both strands, or with complementary sequencing chemistries
The DNA was compared with sequences in the EMBL database using BLASTN and BLASTX26. Transfer RNAs were predicted by tRNAscan-SE27. Potential CDSs were predicted using ORPHEUS28 and GLIMMER29 (both trained on an initial open reading frame set generated by ORPHEUS), and also stop-to-stop prediction; the results were combined. The predicted protein sequences were searched against a non-redundant protein database using WUBLASTP and FASTA. The complete six-frame translation was used to search PROSITE, and the predicted proteins compared against the PFAM30 database of protein domain hidden Markov models. The results of all these analyses were assembled together using the Artemis sequence viewer (K.M.R., unpublished) and used to inform a manual annotation of the sequence and predicted proteins. Annotation was based, wherever possible, on characterized proteins or genes.
References
Blaser, M. J. Epidemiologic and clinical features of Campylobacter jejuni infections. J. Infect. Dis. 176, Suppl 2, S103– S105 (1997).
Nachamkin, I., Allos, B. M. & Ho, T. Campylobacter species and Guillain–Barre syndrome. Clin Microbiol Rev. 11, 555– 567 (1998).
CPHLS. Common gastrointestinal infections, England and Wales. Communicable Dis. Report Weekly 9, 11–13 (1999).
Lobry, J. R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 13, 660–665 (1996).
Moxon, E. R., Rainey, P. B., Nowak, M. A. & Lenski, R. E. Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr. Biol. 4, 24–33 ( 1994).
Hood, D. W. et al. DNA repeats identify novel virulence genes in Haemophilus influenzae. Proc. Natl Acad. Sci. USA 93, 11121–11125 (1996).
Lavitola, A. et al. Intracistronic transcription termination in polysialyltransferase gene (siaD) affects phase variation in Neisseria meningitidis. Mol. Microbiol. 33, 119– 127 (1999).
Alm, R. A. et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397, 176–180 (1999).
Domingo, E., Menendez-Arias, L. & Holland, J. J. RNA virus fitness. Rev. Med. Virol. 7, 87–96 (1997).
Wassenaar, T. M. Toxin production by Campylobacter spp. Clin. Microbiol. Rev. 10, 466-76 (1997).
Wren, B. W. et al. Characterization of a haemolysin from Mycobacterium tuberculosis with homology to a virulence factor of Serpulina hyodysenteriae. Microbiology. 144, 1205– 1211 (1998).
Wood, A., Oldfield, N., O'Dwyer, C. & Ketley, J. Cloning, mutation and distribution of a putative lipopolysaccharide biosynthesis locus in Campylobacter jejuni. Microbiology. 145, 379–388 (1999).
Fry, B., Oldfield, N., Korolik, V., Coloe, P. & Ketley J. in Campylobacter jejuni: Current Status and Future Trends (ed. Nachamkin, I. & M. Blaser, M.) (ASM Press, Washington, DC, in the press).
Guerry, P. et al. Identification and characterization of genes required for posttranslational modification of Campylobacter coli VC167 flagellin. Mol. Microbiol. 19, 369– 378 (1996).
Tomb, J. F. et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539– 547 (1997).
Konkel, M. E., Kim, B. J., Rivera-Amill, V. & Garvis, S. G. Bacterial secreted proteins are required for the internalization of Campylobacter jejuni into cultured mammalian cells. Mol. Microbiol. 32, 691–701 (1999).
Young, G. M., Schmiel, D. H. & Miller, V. L. A new pathway for the secretion of virulence factors by bacteria: the flagellar export apparatus functions as a protein-secretion system. Proc. Natl Acad. Sci. USA 96, 6456 –6461 (1999).
Doig, P., Yao, R., Burr, D. H., Guerry, P. & Trust, T. J. An environmentally regulated pilus-like appendage involved in Campylobacter pathogenesis. Mol. Microbiol. 20, 885–894 (1996).
Yao, R. et al. Isolation of motile and non-motile insertional mutants of Campylobacter jejuni: the role of motility in adherence and invasion of eukaryotic cells. Mol. Microbiol. 14, 883– 893 (1994).
Jones, A. C. et al. A flagellar sheath protein of Helicobacter pylori is identical to HpaA, a putative N-acetylneuraminyllactose-binding hemagglutinin, but is not an adhesin for AGS cells. J. Bacteriol. 179, 5643–5647 (1997).
Yao, R. J., Burr, D. H. & Guerry, P. CheY-mediated modulation of Campylobacter jejuni virulence. Mol. Microbiol. 23, 1021 –1031 (1997).
van Vliet, A. H. M., Wooldridge, K. G. & Ketley, J. M. Iron responsive gene regulation in a Campylobacter jejuni fur mutant. J. Bacteriol. 180, 5291–5298 (1998).
Baillon, M. -L., van Vliet, A., Ketley, J., Constantinidou, C. & Penn, C. An iron-regulated alkyl hydroperoxide reductase (AhpC) confers aerotolerance and oxidative stress resistance to the microaerophilic pathogen Campylobacter jejuni. J. Bacteriol . 181, 4798–4804 ( 1999).
Hughes, N. J., Clayton, C. L., Chalk, P. A. & Kelly, D. J. Helicobacter pylori porCDAB and oorDABC genes encode distinct pyruvate:flavodoxin and 2-oxoglutarate:acceptor oxidoreductases which mediate electron transport to NADP. J. Bacteriol. 180, 1119–1128 (1998).
Bonfield, J. K., Smith, K. F. & Staden, R. A new DNA sequence assembly program. Nucleic Acids Res. 23, 4992–4999 (1995).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403-410 (1990).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 ( 1997).
Frishman, D., Mironov, A., Mewes, H. W. & Gelfand, M. Combining diverse evidence for gene recognition in completely sequenced bacterial genomes. Nucleic Acids Res. 26, 2941– 2947 (1998).
Salzberg, S. L., Delcher, A. L., Kasif, S. & White, O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 26, 544–548 (1998).
Bateman, A. et al. Pfam 3. 1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 27, 260–262 (1999).
Acknowledgements
We would like to thank N. Loman for the Campylobacter Genome Browser; A. Brás, C. Constantinidou, D. Linton, J. Marchant, N. Oldfield and S. Park for assistance with interpretation of sequence data; A. Bateman for CDS clustering; and S. Bowman, D. Kelly, and D. Maskell for a critical reading of the manuscript. Research in the laboratories of B.W.W. and J.M.K. was funded by the BBSRC and sequencing at the Sanger Centre by the Wellcome Trust through its Beowulf Genomics initiative.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Parkhill, J., Wren, B., Mungall, K. et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403, 665–668 (2000). https://doi.org/10.1038/35001088
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/35001088
This article is cited by
-
Identification of Campylobacter jejuni and Campylobacter coli genes contributing to oxidative stress response using TraDIS analysis
BMC Microbiology (2024)
-
High molecular/low acetylated chitosans reduce adhesion of Campylobacter jejuni to host cells by blocking JlpA
Applied Microbiology and Biotechnology (2024)
-
Application of TraDIS to define the core essential genome of Campylobacter jejuni and Campylobacter coli
BMC Microbiology (2023)
-
CamPype: an open-source workflow for automated bacterial whole-genome sequencing analysis focused on Campylobacter
BMC Bioinformatics (2023)
-
Genomic diversity and antimicrobial resistance of Campylobacter spp. from humans and livestock in Nigeria
Journal of Biomedical Science (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
