
Abstract
Originally published as Nature 398, 84—90; 1999
Cbl is an adaptor protein that functions as a negative regulator of many signalling pathways that start from receptors at the cell surface1,2,3,4. The evolutionarily conserved amino-terminal region of Cbl (Cbl-N) binds to phosphorylated tyrosine residues and has cell-transforming activity. Point mutations in Cbl that disrupt its recognition of phosphotyrosine also interfere with its negative regulatory function and, in the case of v-cbl, with its oncogenic potential5. In T cells, Cbl-N binds to the tyrosine-phosphorylated inhibitory site of the protein tyrosine kinase ZAP-706. Here we describe the crystal structure of Cbl-N, both alone and in complex with a phosphopeptide that represents its binding site in ZAP-70. The structures show that Cbl-N is composed of three interacting domains: a four-helix bundle (4H), an EF-hand7 calcium-binding domain, and a divergent SH2 domain8 that was not recognizable from the amino-acid sequence of the protein. The calcium-bound EF hand wedges between the 4H and SH2 domains and roughly determines their relative orientation. In the ligand-occupied structure, the 4H domain packs against the SH2 domain and completes its phosphotyrosine-recognition pocket. Disruption of this binding to ZAP-70 as a result of structure-based mutations in the 4H, EF-hand and SH2 domains confirms that the three domains together form an integrated phosphoprotein-recognition module.
Similar content being viewed by others


Main
Cbl becomes tyrosine-phosphorylated upon engagement of several cell-surface receptors, including the multichain immune receptors, and growth-factor and cytokine receptors1,2,3,4. The amino-acid sequence of Cbl reveals a RING-finger9 domain adjacent to the phosphotyrosine-binding Cbl-N segment, and a carboxy-terminal region with numerous docking sites for SH3- and SH2-containing proteins (Fig. 1). The oncogenic v-Cbl includes only the first 357 residues of Cbl and is a potent transforming protein10. The highly conserved Cbl-N region binds to the receptor for epidermal growth factor (EGFR)11, Syk12 and the negative-regulatory phosphorylation site in ZAP-706. Genetic studies in Caenorhabditis elegans revealed that the Cbl homologue Sli-1 inhibits vulval induction by the EGFR13. Expression of D -Cbl, a Drosophila melanogaster Cbl homologue, disrupts EGFR-regulated development of the R7 photoreceptor14. Cbl also diminishes FcεRI-mediated degranulation in mast cells by inhibiting the tyrosine kinase Syk15. Mutational analysis demonstrates that Cbl-N is central to these functions. To understand better the diverse recognition and regulatory functions of Cbl-N, we have determined its three-dimensional structure.

a, Ribbon diagram of unliganded Cbl-N. The N-terminal 4H domain is coloured yellow, the EF-hand domain green, and the SH2 domain blue. Secondary-structure elements are labelled αA–αD in the 4H domain and by established conventions for the EF-hand and SH2 domains. The bound Ca2+ ion is indicated by a red sphere. Arginine 294 is universally conserved in SH2 domains and participates in phosphotyrosine coordination. b, Diagram of c-Cbl domain structure. The Cbl-N region and adjacent RING finger domain are conserved in all Cbl homologues. The C-terminal region, which contains proline-rich segments and tyrosine phosphorylation sites, is more variable and is completely absent in D-Cbl. A putative leucine zipper has been found near the C terminus of Cbl. c, Aligned sequences of the Cbl-N portion of human c-Cbl, human Cbl-b, Drosophila D-Cbl, and Sli-1. Residues that are identical in at least three of the sequences are shaded yellow. Secondary-structure elements are shown above the sequence and are coloured as in a and b. Black squares indicate residues that coordinate calcium. Red circles mark residues that interact with the bound ZAP-70 peptide. d, Structure-based sequence alignment of Cbl and Lck23 SH2 domains. Seventy structurally equivalent residues are shaded yellow; α-carbons of these seventy residues superimpose with an r.m.s.d. of 1.47 Å. The secondary-structure elements that are present in Lck and other SH2 domains, but not in the Cbl SH2 domain, are indicated by open boxes. e, Superposition of the Cbl SH2 domain (blue) with the Lck SH2 domain (yellow). The structural elements that are absent in the Cbl domain are red.
As shown in Fig. 1, Cbl-N comprises three interacting domains: an N-terminal four-helix bundle (4H), a calcium-binding domain with the EF-hand fold, and an unusual SH2 domain. None of these folding motifs were previously recognized in the amino-acid sequence of Cbl. In spite of the structural and functional similarity of this unusual SH2 domain with other SH2 domains, it shares very little sequence identity (∼11%) with them.
The N-terminal 4H domain contains four long α-helices. Structural comparisons with the DALI16 server show that it has a topology and overall structure similar to many functionally unrelated four-helical proteins, including cytochrome c, interleukin-5 and apolipophorin III. The C and D helices in this domain pack against the adjacent EF-hand domain, and a highly conserved loop connecting the A and B helices contacts the SH2 domain.
The EF-hand motif is similar to those of classical EF-hand proteins such as troponin C and calmodulin. The classical EF-hand fold contains two calcium-binding sites that are formed by the loops connecting the two pairs of E and F helices7. In Cbl, we observe a bound Ca2+ ion only in the more carboxy-terminal E2F2 motif. Although it lacks a canonical calcium-binding sequence pattern, the loop coordinates calcium in a pentagonal–bipyramidal fashion, as in canonical EF-hand proteins (Fig. 2). The paucity of acidic residues in the Cbl loop may be compensated for by interactions with the four-helix bundle, which contributes one of the axial calcium ligands. Glu 164 in helix D coordinates calcium through a bridging water molecule. Typical EF hands use an acidic residue at position 9 in the EF loop to coordinate at this position, sometimes through a bridging water molecule17. The E1F1 loop in Cbl apparently cannot bind calcium: it is one residue shorter than a canonical loop, lacks conserved acidic residues, and does not bind calcium in either of our structures. EF-hand domains occur in several multidomain signalling proteins, often without a clear regulatory function. Phospholipase Cδ contains a calcium-bound EF hand, but it does not interact strongly with other domains in the protein and its function is unknown18. Eps15-homology (EH) domains are essentially EF-hand proteins; some of these domains bind calcium, but not in a way that affects their binding with ligands19. Structures determined for STAT proteins20,21 have revealed an EF-hand-like ‘linker’ domain that makes substantial contact with an adjacent SH2 domain; however, the EF/SH2 interaction in these STAT structures is unlike that in Cbl and the STAT domains do not bind calcium.

The map is a three-fold-averaged SIRAS map, calculated with phases extended to 2.25 Å. The map is contoured at 1.2 σ, and is shown with the refined atomic model. A yellow cross marks the position of the bound calcium ion. The observed pentagonal–bipyramidal coordination is characteristic of EF-hand proteins.
The SH2 domain in Cbl-N retains the general helix–sheet–helix architecture of the SH2 fold (Fig. 1d, e)8, but lacks the secondary β-sheet, comprising β-strands D′, E and F, and also a prominent BG loop. The BG and EF loops in most SH2 domains form a pocket or groove that binds the specificity-determining residues just C-terminal to phosphotyrosine (pY). The phosphotyrosine-binding pocket in Cbl-N is better conserved. The universally conserved arginine (Arg βB5), which makes two hydrogen bonds with the phosphate group, is present in Cbl (Arg 294). In spite of the considerable divergence of the sequence and structure of the Cbl SH2 domain from other SH2 domains, the ZAP-70 phosphopeptide binds as other SH2/phosphopeptide complexes do: the peptide extends across the surface of the domain, roughly perpendicular to the edge of the central β-sheet. The phosphotyrosine residue inserts into a pocket on one side of the sheet, and C-terminal residues are coordinated on the opposite side ( Fig. 3). A portion of the phosphotyrosine pocket is formed by conserved residues in the AB loop of the 4H domain (Fig. 3d). The carbonyl of Pro 81 hydrogen-bonds to a phosphate oxygen through a bridging water molecule, and Pro 82, which is in a cis conformation, packs against the phosphotyrosine-binding BC loop. Additional interactions within the phosphotyrosine pocket are similar to those in other SH2-domain complexes (Fig. 3b). Comparison of the liganded and unliganded structures shows that phosphopeptide binding induces a ‘closure’ of the domains, which creates the intimate association of the 4H domain with the SH2 domain (Fig. 3c). The relative orientations of the 4H domain and EF-hand domain are unchanged, but the SH2 domain rotates by more than 10°, shifting its position by ∼5 Å. The shifted domain packs against helix D and the AB loop in the 4H domain, thereby completing the phosphotyrosine-binding pocket (Fig. 3d). A similar mechanism is employed in the tandem SH2 domains of ZAP-70, where residues of the abutting C-terminal SH2 domain complete the phosphotyrosine-binding pocket of the incomplete N-terminal domain upon binding appropriately spaced phosphotyrosine motifs22.

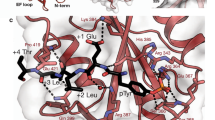
a, Stereo diagram showing an α-carbon trace of the complex. The bound ZAP-70 phosphopeptide is shown in magenta. b, Stereo diagram showing the interactions with the ZAP-70 phosphopeptide. The bound peptide is shown in white. Red spheres represent ordered water molecules that bridge Cbl-N and the bound peptide. Thin blue lines represent hydrogen bonds. In the phosphotyrosine pocket, Tyr 274 in Cbl makes an ‘edge-face’ interaction with the phosphotyrosine ring, and its hydroxyl group hydrogen-bonds to the carbonyl oxygen of Gly 291 in the ZAP-70 peptide. An arginine residue found in this position in most SH2 domains makes an ‘amino–aromatic’ interaction with the phosphotyrosine ring and also hydrogen-bonds with the carbonyl of the pY-1 residue of the bound peptide8. C-terminal to the phosphotyrosine, the proline at position pY+4 in the ZAP-70 peptide binds in a hydrophobic cleft formed by Tyr 307, Phe 336 and Tyr 337, and the glutamic acid residue at pY+3 hydrogen-bonds with the backbone amide of His 320. c, Superposition of the liganded (yellow) and unliganded (blue) Cbl-N structures reveals a shift in the position of the SH2 domain upon phosphopeptide binding. The conformation of the 4H and EF-hand domains is essentially identical in the two structures. In the absence of phosphopeptide, the SH2 domain makes little contact with the 4H domain and its position is likely to vary, as we observe slightly different conformations among the three molecules in the asymmetric unit. Phosphopeptide binding induces a domain ‘closure’, in which the SH2 domain rotates to pack against the helical domain, completing the phosphotyrosine-binding pocket, as in d. d, Molecular surface representation of the Cbl-N domain, coloured by domain. The 4H domain (yellow) forms a portion of the phosphotyrosine-binding pocket. Residues 289–297 of the bound ZAP-70 phosphopeptide are shown as a stick model. The three N-terminal residues in the peptide are disordered and are not included. In the liganded structure, about 1, 200 Å2 of the SH2 domain is buried as a result of interaction with the other two domains; 500 Å2 is buried in the interface with the 4H domain, and 700 Å2 is buried in the interface with the EF hand. The 4H and EF-hand domains share a solvent-excluding interface of ∼800 Å2.
The primary specificity-determining interactions in the Cbl-N /ZAP-70 complex appear to be C-terminal to the phosphotyrosine. In particular, Pro 296 in the ZAP-70 peptide (pY+4) packs in a shallow hydrophobic cleft ( Fig. 3b). This pocket might accept other medium-sized hydrophobic residues as well as proline. A glutamic acid residue at position pY+3 in the peptide also makes specific contacts. In contrast, residues N-terminal to the phosphotyrosine are poorly ordered and we observe no electron density for the first three residues of the ZAP-70 peptide. A phosphopeptide library screen with Cbl-N indicated that the binding motif for the domain could be D(N/D)XpY6. The library varied in the three positions preceding and following the phosphotyrosine, and contained lysine residues at positions C-terminal to pY+3. The Cbl-N structure indicates that a hydrophobic residue at pY+4 is probably needed for high-affinity binding, so predictions made on the basis of the library screen may not be accurate. However, a preference for asparagine at pY-2, as predicted from the screen, is consistent with the observed interactions in the structure. Asp 290 (at pY-2) is poorly ordered, but makes an unexpected hydrogen bond to a phosphate oxygen. We would expect an asparagine residue at this position to make a more favourable interaction. Phosphorylation sites with aspartate or asparagine residues at pY-2 and hydrophobic residues at pY+4 are found in other Cbl-binding partners, including Syk and the EGF receptor.
Several features of the Cbl-N/ZAP-70 complex suggest that the 4H, EF-hand and SH2 domains together form an integrated structure that is crucial for phosphoprotein recognition. As we have discussed, the AB loop of the 4H domain forms a portion of the phosphotyrosine-binding pocket. The EF-hand domain roughly positions the 4H domain with respect to the SH2 domain in a way that seems to require calcium; the calcium-binding site in the EF hand is at the centre of an ∼800 Å2 interface with the 4H domain. Calcium coordination may define the orientation of the E2 and F2 helices, which form the interface with the SH2 domain. In calcium-regulated EF-hand proteins, binding of calcium to the EF loop stabilizes an ‘open’ conformation of the E and F helices that is required for interaction with target helical peptides7. In our structures, the calcium-bound E2F2 motif adopts an open conformation, and helix αN, which connects the EF-hand and SH2 domains in the primary structure, is positioned between the ends of the E2 and F2 helices in a position similar to that occupied by bound helical peptides in calmodulin structures.
To test whether the three domains form an integrated recognition module, we mutated key residues in each domain and compared the ability of wild-type and mutant proteins to precipitate ZAP-70 from activated Jurkat T-cell lysates. In the SH2 domain, substitution of lysine for the universally conserved ‘FLVRES’ arginine (Arg 294) disrupts the interaction of Cbl-N with ZAP-70, as does mutation of Gly 306 to glutamic acid (Fig. 4). This substitution corresponds to a loss-of-function mutation discovered in Sli-113 and disrupts the phosphotyrosine-binding function of Cbl and the transforming ability of v-cbl3,5. The mutation is in strand βC of the SH2 domain, near the phosphotyrosine pocket. The structure suggests that a glutamic acid at this position could form a buried salt bridge with Arg 294, preventing its interaction with phosphotyrosine. We disrupted the calcium-binding site in the EF hand by mutating acidic residues that participate in calcium coordination (Fig. 4). Mutation of Glu 240 to serine prevented the fusion protein of glutathione-S-transferase with Cbl-N (GST–Cbl-N) from interacting with ZAP-70, and mutation of Asp 229 to glutamine markedly weakened this interaction. In the 4H domain, we substituted aspartate for Ser 80 and alanine for Pro 82 to test the importance of the contribution of the AB loop to the phosphotyrosine-binding pocket. We detected no precipitation of phosphorylated ZAP-70 for either substitution. On the basis of the structure and our mutagenesis results, we conclude that the three domains together form the functional phosphoprotein-binding unit, and that calcium coordination plays a key structural role in phosphoprotein recognition.

Jurkat T cells were left untreated (−) or stimulated by anti-TCR crosslinking using OKT3 antibody (+). Lysates from resting or activated cells were probed with wild-type or mutant GST–Cbl-N bound to glutathione beads and the associated proteins were resolved by SDS–PAGE and immunoblotted for the presence of tyrosine-phosphorylated proteins using the anti-phosphotyrosine antibody RC20H (a). The same blot was stripped and re-probed with an anti-ZAP-70 antibody (b). Mutation of Gly 306 to Glu (corresponding to the loss-of-function mutation in Sli-1) or Arg 294 to Lys within the SH2 domain disrupt association of Cbl-N with ZAP-70. Mutations in the calcium-binding E2F2 loop, including Asp 229 to Gln and Glu 240 to Ser diminish recognition of ZAP-70 by Cbl-N. Substitutions for Ser 80 and Pro 82 in the AB loop of the 4H domain also prevent ZAP-70 precipitation.
Whether Cbl function is regulated in vivo by changes in intracellular Ca2+ concentration remains to be investigated. The affinity of Cbl for calcium may be sufficiently high that it is constitutively calcium-bound; no calcium was added to the unbound Cbl-N and the crystallization buffer included citrate, a weak Ca2+-chelator. Addition of 1 mM EGTA to activated Jurkat lysates does not prevent precipitation of ZAP-70 by GST–Cbl-N (data not shown), indicating that calcium probably binds with high affinity. Thus, our in vitro results obtained with the isolated Cbl-N domain suggest that the calcium site is unlikely to serve a regulatory role. However, its regulatory potential depends upon its affinity for calcium in the cellular milieu; interactions with other domains in Cbl or with other proteins may alter its affinity for calcium. The architectural complexity of Cbl-N, in comparison with typical SH2 or PTB domains, suggests that it has a capacity for an additional binding or regulatory function.
Methods
Expression, purification and crystallization. Cbl-N was produced as a GST-fusion protein in E. coli strain DH5α using the expression plasmid pGEX-4T-3 (Pharmacia). Cleared cell lysates were incubated overnight with glutathione–agarose beads. After extensive washing with PBS, the desired Cbl-N fragment was digested from the beads with thrombin (using a molar ratio of about 1:500) in 50 mM Tris, pH 7.4, 200 mM NaCl, and 2.5 mM calcium chloride. The digested Cbl-N was recovered and further purified by affinity chromatography on phosphotyrosine–Sepharose23. The purified protein was dialysed exhaustively against storage buffer (20 mM HEPES, pH 7.0, 200 mM NaCl, 2mM DTT) and concentrated to 5 mg ml−1 for crystallization in a Centricon (Amicon). The purified protein includes residues 25–351 of human Cbl, plus two residues (Gly-Ser) from the GST fusion.
Cbl-N crystals were obtained by combining 25 µl protein and 25 µl precipitant solution (20% PEG 4000, 100 mM sodium citrate, pH 5.6, 200 mM ammonium acetate) in a sealed glass depression plate at 22 °C. The crystals were maintained in a cryo-stabilization buffer (22% PEG 4000, 100 mM sodium citrate, pH 5.6, 200 mM ammonium acetate, 200 mM NaCl, 20% glycerol) for at least 2 h before flash-freezing by plunging into liquid nitrogen. For co-crystallization, a 2-fold molar excess of the ZAP-70 pTyr-292 peptide TLNSDGpYTPEPA was added to Cbl-N at 5 mg ml−1 in storage buffer. Crystals were grown in hanging drops at 22 °C by mixing 2 µl protein/peptide solution with 2 µl well solution containing 20% PEG 8000, 0.2 M calcium acetate, 2 mM DTT, and 0.1 M sodium cacodylate, pH 6.1. Cbl/ZAP-70 co-crystals were briefly dunked in a stabilizing buffer containing the well solution plus 20% glycerol, and flash- frozen in liquid nitrogen. All diffraction data were obtained by using a Quantum-4 CCD detector (ADSC) on the F1 beam line at CHESS (Table 1) at −165 °C. Diffraction images were indexed, integrated and scaled using DENZO and SCALEPACK24 (Cbl/ZAP-70 complex) or MOSFLM25 (unliganded structure).
Cbl-N structure determination. The unliganded Cbl-N structure was determined by SIRAS using a methyl mercury nitrate derivative (1 mM, overnight soak). Fifteen mercury sites were located by using difference-Patterson and difference-Fourier methods using the CCP425 program suite and the programs PATSOL (L. Tong) and HEAVY (T. Terwilliger). Structure-factor phases were calculated using MLPHARE25, and improved with solvent-flipping in DM25. An electron density map calculated using these phases at 2.9 Å resolution revealed clear main-chain density and substantial side-chain detail. With the aid of skeletonization using BONES26, it was possible to trace the path of the main chain for one of the three molecules in the asymmetric unit (molecule B). Density for the other two molecules was broken and hard to interpret. The skeleton for molecule B was used to generate a molecular envelope for NCS averaging. Initial rotation matrices describing the relative orientations of molecules A, B and C were calculated from the heavy-atom sites (mercury bound to five sites in each of the three molecules). NCS averaging with DM25 was used to improve phases at 2.7 Å resolution, and then extend phases to 2.2 Å, the limit of the native data set. The resulting electron density map was readily interpretable (Fig. 2). A model including residues 47–351 of each of the three molecules was built using the graphics program O26. No electron density is seen for 24 residues at the amino terminus. The model was refined using simulated annealing and positional refinement in X-PLOR27, with tight NCS restraints. The model includes 669 water molecules, which were positioned by using the program ARP (V.Lamzin). An overall thermal B factor and tightly restrained individual B factors were refined, and a bulk-solvent model was incorporated. Crystallographic R values and stereochemical parameters are presented in Table 1.
The structure of the Cbl-N/ZAP70 phosphopeptide complex was determined by molecular replacement with the program AmoRe28. Use of the complete unliganded Cbl-N structure as a search model yielded clear rotation and translation peaks, and maps phased with the appropriately positioned model revealed strong electron density for the 4H and EF-hand domains, but no interpretable density for the SH2 domain. The model was therefore broken into two fragments (residues 47–265 and residues 266–351), and the rotation and translation searches were repeated with each fragment. The 4H/EF-hand fragment yielded a solution essentially identical to that from the intact model. The SH2 domain was then positioned using translation searches conducted in the context of the appropriately positioned 4H/EF-hand fragment. After rigid-body and positional refinement using X-PLOR27, electron density maps calculated with the combined model revealed clear density for all domains, and readily interpretable density for the bound ZAP-70 phosphopeptide. After construction of the peptide, the structure was refined with iterative cycles of manual refitting and simulated annealing and positional refinement in X-PLOR (Table 1). Restrained individual temperature factors were refined. The model includes residues 47–351 of c-Cbl, residues 289–297 of ZAP-70, and 358 water molecules.
Illustrations. Figure 1awas prepared with MOLSCRIPT29, Fig. 2 with program O26, and Figs 1e and 3 with GRASP30.
References
Thien, C. B. & Langdon, W. Y. c-Cbl: a regulator of T cell receptor-mediated signalling. Immunol. Cell Biol. 76 , 473–482 (1998).
Liu, Y. C. & Altman, A. Cbl: Complex formation and functional implications. Cell Signal. 10, 377– 385 (1998).
Miyake, S.et al. The Cbl proto-oncogene product: From enigmatic oncogene to center stage of signal transduction. Crit. Rev. Oncogen. 8, 189–218 (1998).
Wange, R. L. & Samelson, L. E. Complex complexes: signaling at the TCR. Immunity 5, 197– 205 (1996).
Thien, C. B. & Langdon, W. Y. EGF receptor binding and transformation by v-cbl is ablated by the introduction of a loss-of-function mutation from the Caenorhabditis elegans sli-1 gene. Oncogene 14, 2239–2249 ( 1997).
Lupher, M. L. J, Songyang, Z., Shoelson, S. E., Cantley, L. C. & Band, H. The Cbl phosphotyrosine-binding domain selects a D(N/D)XpY motif and binds to the Tyr292 negative regulatory phosphorylation site of ZAP-70. J. Biol. Chem. 272, 33140 –33144 (1997).
Ikura, M. Calcium binding and conformational response in EF-hand proteins. Trends Biochem. Sci. 21, 14–17 (1996).
Kuriyan, J. & Cowburn, D. Modular peptide recognition domains in eukaryotic signaling. Annu. Rev. Biophys. Biomol. Struct. 26, 259–288 (1997).
Saurin, A. J., Borden, K. L., Boddy, M. N. & Freemont, P. S. Does this have a familiar RING? Trends Biochem. Sci. 21, 208–214 (1996).
Blake, T. J., Shapiro, M., Morse, H. C. & Langdon, W. Y. The sequences of the human and mouse c-cbl proto-oncogenes show v- cbl was generated by a large truncation encompassing a proline-rich domain and a leucine zipper-like motif. Oncogene 6, 653–657 (1991).
Galisteo, M. L., Dikic, I., Batzer, A. G., Langdon, W. Y. & Schlessinger, J. Tyrosine phosphorylation of the c-cbl proto-oncogene protein product and association with epidermal growth factor (EGF) receptor upon EGF stimulation. J. Biol. Chem. 270, 20242–20245 (1995).
Deckert, M., Elly, C., Altman, A. & Liu, Y. C. Coordinated regulation of the tyrosine phosphorylation of Cbl by Fyn and Syk tyrosine kinases. J. Biol. Chem. 273, 8867–8874 (1998).
Yoon, C. H., Lee, J., Jongeward, G. D. & Sternberg, P. W. Similarity of sli-1, a regulator of vulval development in C. elegans , to the mammalian proto-oncogene c-cbl. Science 269, 1102–1105 (1995).
Meisner, H. etal. Interactions of Drosophila Cbl with epidermal growth factor receptors and role of Cbl in R7 photoreceptor cell development. Mol. Cell Biol. 17, 2217–2225 ( 1997).
Ota, Y. & Samelson, L. E. The product of the proto-oncogene c-cbl: a negative regulator of the Syk tyrosine kinase. Science 276, 418–420 ( 1997).
Holm, L. & Sander, C. Dali anetwork tool for protein structure comparison. Trends Biochem. Sci. 20, 478– 480 (1995).
Kretsinger, R. H. EF-hands embrace. Nature Struct. Biol. 4, 514–516 (1997).
Essen, L. O., Perisic, O., Cheung, R., Katan, M. & Williams, R. L. Crystal structure of a mammalian phosphoinositide-specific phospholipase Cδ. Nature 380, 595– 602 (1996).
de Beer, T., Carter, R. E., Lobel-Rice, K. E., Sorkin, A. & Overduin, M. Structure and Asn-Pro-Phe binding pocket of the Eps15 homology domain. Science 281, 1357–1360 (1998).
Becker, S., Groner, B. & Muller, C. W. Three-dimensional structure of the Stat3β homodimer bound to DNA. Nature 394, 145– 151 (1998).
Chen, X.et al. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell 93, 827–839 (1998).
Hatada, M. H. etal. Molecular basis for the interaction of the protein tyrosine kinase ZAP-70 with the T-cell receptor. Nature 377, 32– 38 (1995).
Eck, M. J., Shoelson, S. E. & Harrison, S. C. Recognition of a high-affinity phosphotyrosyl peptide by the Src homology-2 domain of p56lck. Nature 362, 87–91 (1993).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 ( 1997).
Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50, 760– 776 (1994).
Jones, T. A., Zhou, J. Y., Cowan, S. W. & Kjeldgaard, M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 (1991 ).
Brunger, A. X-PLOR Version 3.0: A System for Crystallography and NMR (Yale University Press, New Haven, (1992).
Navaza, J. (ed. AMoRe: A New Package for Molecular Replacement (SERC, Daresbury, UK, (1992).
Kraulis, P. J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24, 946 –950 (1991).
Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281–296 ( 1991).
Acknowledgements
We thank C. Dahl for synthesis and purification of the ZAP-70 phosphopeptide, the staff at MacCHESS for assistance with data collection, and S. Harrison and T. Roberts for comments on the manuscript. M.J.E. is a recipient of a Burroughs–Wellcome Career Award in the Biomedical Sciences. Diffraction data were recorded at the Cornell High Energy Synchrotron Source (CHESS), which is supported by grants from the NSF and NIH.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Meng, W., Sawasdikosol, S., Burakoff, S. et al. Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature 402 (Suppl 6763), 29–34 (1999). https://doi.org/10.1038/31000000
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/31000000
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
