
Abstract
A total of 28 children and nine adults with relapsed T-ALL were analyzed for the configuration of their T-cell receptor (TCR) and TAL1 genes at diagnosis and relapse to evaluate their stability throughout the disease course. A total of 150 clonal TCR and TAL1 gene rearrangements were identified in the 37 patients at diagnosis. In 65% of cases all rearrangements and in 27% of cases most rearrangements found at diagnosis were preserved at relapse. Two children with unusually late T-ALL recurrences displayed completely different TCR gene rearrangement sequences between diagnosis and relapse. This indicates that a proportion of very late T-ALL recurrences might represent second T-ALL. Specifically, 88% of clonal rearrangements identified at diagnosis in truly relapsed T-ALL were preserved at relapse. This is significantly higher as compared to previously studied precursor-B-ALL (∼70%). Thus, from biological point of view, immunogenotype of T-ALL is more stable as compared with precursor-B-ALL. The overall stability of TCR gene rearrangements was higher in adult T-ALL (97%) than in childhood T-ALL (86%). Based on the stability of TCR gene rearrangements, we propose a strategy for PCR target selection (TCRD+TAL1 → TCRB → TCRG), which probably allows reliable minimal residual disease detection in all T-ALL patients.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
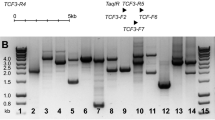

References
Szczepański T, Orfao A, van der Velden VHJ, San Miguel JF, van Dongen JJM . Minimal residual disease in leukaemia patients. Lancet Oncol 2001; 2: 409–417.
Cave H, van der Werff ten Bosch J, Suciu S, Guidal C, Waterkeyn C, Otten J et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. N Engl J Med 1998; 339: 591–598.
Van Dongen JJM, Seriu T, Panzer-Grümayer ER, Biondi A, Pongers-Willemse MJ, Corral L et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 1998; 352: 1731–1738.
Coustan-Smith E, Sancho J, Hancock ML, Boyett JM, Behm FG, Raimondi SC et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood 2000; 96: 2691–2696.
Nyvold C, Madsen HO, Ryder LP, Seyfarth J, Svejgaard A, Clausen N et al. Precise quantification of minimal residual disease at day 29 allows identification of children with acute lymphoblastic leukemia and an excellent outcome. Blood 2002; 99: 1253–1258.
Willemse MJ, Seriu T, Hettinger K, d’Aniello E, Hop WCJ, Panzer-Grümayer ER et al. Detection of minimal residual disease identifies differences in treatment response between T-ALL and precursor-B-ALL. Blood 2002; 99: 4386–4393.
Pui CH, Campana D . New definition of remission in childhood acute lymphoblastic leukemia. Leukemia 2000; 14: 783–785.
Szczepański T, Flohr T, van der Velden VHJ, Bartram CR, van Dongen JJM . Molecular monitoring of residual disease using antigen receptor genes in childhood acute lymphoblastic leukaemia. Best Pract Res Clin Haematol 2002; 15: 37–57.
Pongers-Willemse MJ, Seriu T, Stolz F, d'Aniello E, Gameiro P, Pisa P et al. Primers and protocols for standardized MRD detection in ALL using immunoglobulin and T cell receptor gene rearrangements and TAL1 deletions as PCR targets. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia 1999; 13: 110–118.
Verhagen OJHM, Willemse MJ, Breunis WB, Wijkhuijs AJM, Jacobs DCH, Joosten SA et al. Application of germline IGH probes in real-time quantitative PCR for the detection of minimal residual disease in acute lymphoblastic leukemia. Leukemia 2000; 14: 1426–1435.
Brüggemann M, Droese J, Bolz I, Luth P, Pott C, von Neuhoff N et al. Improved assessment of minimal residual disease in B cell malignancies using fluorogenic consensus probes for real-time quantitative PCR. Leukemia 2000; 14: 1419–1425.
Donovan JW, Ladetto M, Zou G, Neuberg D, Poor C, Bowers D et al. Immunoglobulin heavy-chain consensus probes for real-time PCR quantification of residual disease in acute lymphoblastic leukemia. Blood 2000; 95: 2651–2658.
Van der Velden VHJ, Willemse MJ, van der Schoot CE, van Wering ER, van Dongen JJM . Immunoglobulin kappa deleting element rearrangements in precursor-B acute lymphoblastic leukemia are stable targets for detection of minimal residual disease by real-time quantitative PCR. Leukemia 2002; 16: 928–936.
Van der Velden VHJ, Wijkhuijs JM, Jacobs DCH, van Wering ER, van Dongen JJM . T cell receptor gamma gene rearrangements as targets for detection of minimal residual disease in acute lymphoblastic leukemia by real-time quantitative PCR analysis. Leukemia 2002; 16: 1372–1380.
Van der Velden VHJ, Jacobs DCH, Wijkhuijs AJM, Comans-Bitter WM, Willemse MJ, Hählen K et al. Minimal residual disease levels in bone marrow and peripheral blood are comparable in children with T cell acute lymphoblastic leukemia (ALL), but not in precursor-B-ALL. Leukemia 2002; 16: 1432–1436.
Szczepański T, Pongers-Willemse MJ, Langerak AW, van Dongen JJM . Unusual immunoglobulin and T-cell receptor gene rearrangement patterns in acute lymphoblastic leukemias. Curr Top Microbiol Immunol 1999; 246: 205–215.
De Haas V, Verhagen OJ, von dem Borne AE, Kroes W, van den Berg H, van der Schoot CE . Quantification of minimal residual disease in children with oligoclonal B-precursor acute lymphoblastic leukemia indicates that the clones that grow out during relapse already have the slowest rate of reduction during induction therapy. Leukemia 2001; 15: 134–140.
Szczepański T, Willemse MJ, Brinkhof B, van Wering ER, van der Burg M, van Dongen JJM . Comparative analysis of Ig and TCR gene rearrangements at diagnosis and at relapse of childhood precursor-B-ALL provides improved strategies for selection of stable PCR targets for monitoring of minimal residual disease. Blood 2002; 99: 2315–2323.
Beishuizen A, Verhoeven MA, van Wering ER, Hählen K, Hooijkaas H, van Dongen JJM . Analysis of Ig and T-cell receptor genes in 40 childhood acute lymphoblastic leukemias at diagnosis and subsequent relapse: implications for the detection of minimal residual disease by polymerase chain reaction analysis. Blood 1994; 83: 2238–2247.
Taylor JJ, Rowe D, Kylefjord H, Chessells J, Katz F, Proctor SJ et al. Characterisation of non-concordance in the T-cell receptor gamma chain genes at presentation and clinical relapse in acute lymphoblastic leukemia. Leukemia 1994; 8: 60–66.
Baruchel A, Cayuela JM, MacIntyre E, Berger R, Sigaux F . Assessment of clonal evolution at Ig/TCR loci in acute lymphoblastic leukaemia by single-strand conformation polymorphism studies and highly resolutive PCR derived methods: implication for a general strategy of minimal residual disease detection. Br J Haematol 1995; 90: 85–93.
LoNigro L, Cazzaniga G, DiCataldo A, Pannunzio A, DAniello E, Masera G et al. Clonal stability in children with acute lymphoblastic leukemia (ALL) who relapsed five or more years after diagnosis. Leukemia 1999; 13: 190–195.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR et al. Proposals for the classification of the acute leukaemias. French–American–British (FAB) co-operative group. Br J Haematol 1976; 33: 451–458.
Van Dongen JJM, Adriaansen HJ, Hooijkaas H . Immunophenotyping of leukaemias and non-Hodgkin's lymphomas. Immunological markers and their CD codes. Neth J Med 1988; 33: 298–314.
Szczepański T, Willemse MJ, Kamps WA, van Wering ER, Langerak AW, van Dongen JJM . Molecular discrimination between relapsed and secondary acute lymphoblastic leukemia – proposal for an easy strategy. Med Pediatr Oncol 2001; 36: 352–358.
Van Dongen JJM, Wolvers-Tettero ILM . Analysis of immunoglobulin and T cell receptor genes. Part I: basic and technical aspects. Clin Chim Acta 1991; 198: 1–91.
Langerak AW, Wolvers-Tettero ILM, van Dongen JJM . Detection of T cell receptor beta (TCRB) gene rearrangement patterns in T cell malignancies by Southern blot analysis. Leukemia 1999; 13: 965–974.
Breit TM, Wolvers-Tettero ILM, Beishuizen A, Verhoeven M-AJ, van Wering ER, van Dongen JJM . Southern blot patterns, frequencies and junctional diversity of T-cell receptor δ gene rearrangements in acute lymphoblastic leukemia. Blood 1993; 82: 3063–3074.
Quertermous T, Strauss WM, Van Dongen JJM, Seidman JG . Human T cell gamma chain joining regions and T cell development. J Immunol 1987; 138: 2687–2690.
Moreau EJ, Langerak AW, van Gastel-Mol EJ, Wolvers-Tettero ILM, Zhan M, Zhou Q et al. Easy detection of all T cell receptor gamma (TCRG) gene rearrangements by Southern blot analysis: recommendations for optimal results. Leukemia 1999; 13: 1620–1626.
Szczepański T, Langerak AW, Willemse MJ, Wolvers-Tettero ILM, van Wering ER, van Dongen JJM . T cell receptor gamma (TCRG) gene rearrangements in T cell acute lymphoblastic leukemia reflect ‘end-stage’ recombinations: implications for minimal residual disease monitoring. Leukemia 2000; 14: 1208–1214.
Szczepański T, Langerak AW, Wolvers-Tettero ILM, Ossenkoppele GJ, Verhoef G, Stul M et al. Immunoglobulin and T cell receptor gene rearrangement patterns in acute lymphoblastic leukemia are less mature in adults than in children: implications for selection of PCR targets for detection of minimal residual disease. Leukemia 1998; 12: 1081–1088.
Langerak AW, Szczepański T, van der Burg M, Wolvers-Tettero ILM, van Dongen JJM . Heteroduplex PCR analysis of rearranged T cell receptor genes for clonality assessment in suspect T cell proliferations. Leukemia 1997; 11: 2192–2199.
Ghali DW, Panzer S, Fischer S, Argyriou-Tirita A, Haas OA, Kovar H et al. Heterogeneity of the T-cell receptor delta gene indicating subclone formation in acute precursor B-cell leukemias. Blood 1995; 85: 2795–2801.
Szczepański T, Pongers-Willemse MJ, Langerak AW, Harts WA, Wijkhuijs JM, van Wering ER et al. Ig heavy chain gene rearrangements in T-cell acute lymphoblastic leukemia exhibit predominant DH6-19 and DH7-27 gene usage, can result in complete V–D–J rearrangements, and are rare in T-cell receptor αβ lineage. Blood 1999; 93: 4079–4085.
Lefranc MP, Giudicelli V, Ginestoux C, Bodmer J, W Müller, Bontrop R et al. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res 1999; 27: 209–212.
Breit TM, Van Dongen JJ . Unravelling human T-cell receptor junctional region sequences. Thymus 1994; 22: 177–199.
Pui CH, Behm FG, Singh B, Schell MJ, Williams DL, Rivera GK et al. Heterogeneity of presenting features and their relation to treatment outcome in 120 children with T-cell acute lymphoblastic leukemia. Blood 1990; 75: 174–179.
Hoelzer D, Gokbuget N, Ottmann O, Pui CH, Relling MV, Appelbaum FR et al. Acute lymphoblastic leukemia. Hematology (Am Soc Hematol Educ Program) 2002; 162–192.
Kamps WA, Bokkerink JP, Hakvoort-Cammel FG, Veerman AJ, Weening RS, van Wering ER et al. BFM-oriented treatment for children with acute lymphoblastic leukemia without cranial irradiation and treatment reduction for standard risk patients: results of DCLSG protocol ALL-8 (1991–1996). Leukemia 2002; 16: 1099–1111.
Amylon MD, Shuster J, Pullen J, Berard C, Link MP, Wharam M et al. Intensive high-dose asparaginase consolidation improves survival for pediatric patients with T cell acute lymphoblastic leukemia and advanced stage lymphoblastic lymphoma: a Pediatric Oncology Group study. Leukemia 1999; 13: 335–342.
Hunger SP, Sklar J, Link MP . Acute lymphoblastic leukemia occurring as a second malignant neoplasm in childhood: report of three cases and review of the literature. J Clin Oncol 1992; 10: 156–163.
Dawson L, Slater R, Hagemeijer A, Langerak AW, Willemze R, Kluin-Nelemans JC . Secondary T-acute lymphoblastic leukaemia mimicking blast crisis in chronic myeloid leukaemia. Br J Haematol 1999; 106: 104–106.
Liso V, Specchia G, Pannunzio A, Mestice A, Palumbo G, Biondi A . T-cell acute lymphoblastic leukemia occurring in a patient with acute promyelocytic leukemia. Haematologica 1998; 83: 471–473.
Kaplinsky C, Frisch A, Cohen IJ, Goshen Y, Jaber L, Yaniv I et al. T-cell acute lymphoblastic leukemia following therapy of rhabdomyosarcoma. Med Pediatr Oncol 1992; 20: 229–231.
Perotti D, Sozzi G, Ferrari A, Casanova M, Gambirasio F, Mondini P et al. Cytogenetic and molecular characterization of T-cell acute lymphoblastic leukemia as a second tumor after anaplastic large-cell lymphoma in a boy. Haematologica 1999; 84: 554–557.
Van Dongen JJM, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia 1999; 13: 1901–1928.
Breit TM, Beishuizen A, Ludwig WD, Mol EJ, Adriaansen HJ, van Wering ER et al. tal-1 deletions in T-cell acute lymphoblastic leukemia as PCR target for detection of minimal residual disease. Leukemia 1993; 7: 2004–2011.
Bash RO, Crist WM, Shuster JJ, Link MP, Amylon M, Pullen J et al. Clinical features and outcome of T-cell acute lymphoblastic leukemia in childhood with respect to alterations at the TAL1 locus: a Pediatric Oncology Group study. Blood 1993; 81: 2110–2117.
Breit TM, Verschuren MCM, Wolvers-Tettero ILM, van Gastel-Mol EJ, Hählen K, van Dongen JJM . Human T cell leukemias with continuous V(D)J recombinase activity for TCR-delta gene deletion. J Immunol 1997; 159: 4341–4349.
Van Wering ER, van der Linden-Schrever BEM, van der Velden VHJ, Szczepański T, van Dongen JJM . T lymphocytes in bone marrow samples of children with acute lymphoblastic leukemia during and after chemotherapy might hamper PCR-based minimal residual disease studies. Leukemia 2001; 15: 1031–1033.
Van Dongen JJM, Quertermous T, Bartram CR, Gold DP, Wolvers-Tettero ILM, Comans-Bitter WM et al. T cell receptor-CD3 complex during early T cell differentiation. Analysis of immature T cell acute lymphoblastic leukemias (T-ALL) at DNA, RNA, and cell membrane level. J Immunol 1987; 138: 1260–1269.
Blom B, Verschuren MC, Heemskerk MH, Bakker AQ, van Gastel-Mol EJ, Wolvers-Tettero IL et al. TCR gene rearrangements and expression of the pre-T cell receptor complex during human T-cell differentiation. Blood 1999; 93: 3033–3043.
Acknowledgements
We are grateful to Professor Dr R Benner and Professor Dr D Sońta-Jakimczyk for their continuous support, and Mrs WM Comans-Bitter for preparation of the figures.
We thank the Board and the Clinicians of the Dutch Childhood Oncology Group for kindly providing T-ALL cell samples.
This study was supported by the Dutch Cancer Foundation/Koningin Wilhelmina Fonds (Grants SNWLK 97-1567 and SNWLK 2000-2268).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Szczepański, T., Velden, V., Raff, T. et al. Comparative analysis of T-cell receptor gene rearrangements at diagnosis and relapse of T-cell acute lymphoblastic leukemia (T-ALL) shows high stability of clonal markers for monitoring of minimal residual disease and reveals the occurrence of second T-ALL. Leukemia 17, 2149–2156 (2003). https://doi.org/10.1038/sj.leu.2403081
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.leu.2403081
Keywords
This article is cited by
-
Utility of Measurable Residual Disease (MRD) Assessment in Mantle Cell Lymphoma
Current Treatment Options in Oncology (2023)
-
Is Next-Generation Sequencing the way to go for Residual Disease Monitoring in Acute Lymphoblastic Leukemia?
Molecular Diagnosis & Therapy (2017)
-
Prognostic value and clinical significance of TCR rearrangements for MRD monitoring in ALL patients
Comparative Clinical Pathology (2017)
-
Very early/early relapses of acute lymphoblastic leukemia show unexpected changes of clonal markers and high heterogeneity in response to initial and relapse treatment
Leukemia (2011)
