
Abstract
Using addictive drugs can evolve from controlled social use into the compulsive relapsing disorder that characterizes addiction. This transition to addiction results from genetic, developmental, and sociological vulnerabilities, combined with pharmacologically induced plasticity in brain circuitry that strengthens learned drug-associated behaviors at the expense of adaptive responding for natural rewards. Advances over the last decade have identified the brain circuits most vulnerable to drug-induced changes, as well as many associated molecular and morphological underpinnings. This growing knowledge has contributed to an expanded understanding of how drugs usurp normal learning circuitry to create the pathology of addiction, as evidenced by involuntary activation of reward circuits in response to drug-associated cues and simultaneous reports of drug craving. This new understanding provides unprecedented potential opportunities for novel pharmacotherapeutic targets in treating addiction. There appears to be plasticity associated with the addiction phenomenon in general as well as changes produced by addiction to a specific class of addicting drugs. These findings also provide the basis for the current understanding of addiction as a chronic, relapsing disease of the brain with changes that persist long after the last use of the drug. Here, we describe the neuroplasticity in brain circuits and cell function induced by addictive drugs that is thought to underlie the compulsions to resume drug-taking, and discuss how this knowledge is impelling exploration and testing of novel addiction therapies.
Similar content being viewed by others
Main
Drug addiction is traditionally underappreciated as a disease rooted in neuropathology (O'Brien, 2003). The perspective that drug-dependent individuals should simply disengage from the self-destructive behaviors that are driven by addiction has distracted society from treating drug abuse as a chronic medical disorder. The last 20 years of research has made it clear that addiction to drugs is based on pathological changes in brain function produced by repeated pharmacological insult to the brain circuits that regulate how a person interprets and behaviorally responds to motivationally relevant stimuli. Thus, addictive drugs strongly interact with and change the brain circuits that permit us to learn about and behaviorally adapt to important environmental stimuli, whether it be how to best approach rewards such as food or sex, or to avoid dangerous situations (Kelley, 2004; Everitt and Robbins, 2005). By changing motivational circuitry, addictive drugs impair the development of behavioral strategies towards biological stimuli in favor of progressively greater orientation of behavior towards drug-seeking and drug-taking strategies (Kalivas and Volkow, 2005). Importantly, these changes are long-lasting and, at present, not readily reversed (Hyman et al, 2006).
The modern definition of addiction was first established in DSM IIIR by the American Psychiatric Association in 1987 (APA, 1987). The clinical picture is marked by compulsive drug use that the individual cannot fully control. Tolerance and withdrawal symptoms may be present, but they do not necessarily signal addiction. Rather, the essential elements consist of persistent and recurrent drug-seeking behavior at the expense of pursuit of normal rewards. The definition assumed, without clear evidence in 1987, that there is a ‘core’ addiction syndrome that pharmacologically diverse drugs of abuse can produce. In this review, we marshal currently available evidence to describe this core syndrome as a neuropathology in the molecular and circuitry underpinnings of motivated behavior. From this perspective, addiction is a pathology in mechanisms of brain neuroplasticity that are used to establish the adaptive hierarchy of behaviors that ensure survival. Thus, enduring drug-induced neuroplasticity establishes a maladaptive orientation to the environment that manifests as the two cardinal features of addiction, (1) impaired ability to regulate the drive to obtain and use drugs (ie, relapse), and (2) reduced drive to obtain natural rewards.
NEUROPLASTICITY AND THE STAGES OF ADDICTION
For this review, neuroplasticity will be operationally parsed into two categories: first, relatively transient changes in neuronal function that continue for hours up to weeks of drug abstinence, and second, relatively stable changes lasting from weeks to being relatively permanent changes. Transient neuroplasticity corresponds to the necessary changes that are antecedent to developing a new behavior, whereas stable neuroplasticity corresponds to the stable information that is retrieved to guide the execution of learned behavior. For addiction, these stages are generally described as developing addiction (ie, learning to become addicted), and a relative stable state of high vulnerability to relapse after stopping drug-taking. The development of addiction is typically achieved through repeated social use of the drug, and involves many relatively short-lived changes in brain chemistry and physiology based largely on the molecular pharmacology of the drug itself (Nestler, 2005). In Figure 1a, this stage is referred to as social use. The second stage is wrought by repeated drug insults and is based on enduring changes in the synaptic physiology of brain circuits regulating cognitive and emotional responding to important environmental stimuli. This is illustrated in Figure 1a as two phases of relapse. The first phase of relapse is defined as regulated relapse, the second as compulsive relapse. Regulated relapse refers to a relatively declarative decision-making process whereby the addict consciously decides to relapse. For example, the individual may make a choice between helping their child with homework or drinking a glass of wine. In this stage, the addict often makes the socially appropriate choice. In compulsive relapse, the addict is not making a conscious choice. For example, although helping their child with homework may have been an agenda item for the evening, exposure to various environmental cues or stressors that the individual has associated with repeated drug use activates drug-seeking, and a conscious decision is never made, the person automatically relapses.

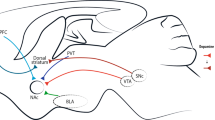
Illustration of the relationship between neuroplasticity, motivated learning, brain circuitry, and the stages of addiction. (a) The phases of addiction from the development of addiction (social use) to vulnerability to relapse (transitioning from regulated to compulsive). Also shown is the proposed use of pharmacotherapeutics and pharmacology and behavioral interventions. (b) Mapping normal motivated learning processes and the relevant dopaminergic and glutamatergic circuitry onto the stages of addiction.
Obviously, compulsive relapse is a more severe phase that, as we shall see, contains potential neuropathological targets for developing pharmacotherapeutic interventions. As illustrated in Figure 1, we will propose towards the end of this review that based on the neuropathology of compulsive relapse being a form of stable neuroplasticity, a primary role of psychopharmacology in treating addiction is to develop drugs that promote regulated over compulsive relapse. In other words, among the most valuable pharmacotherapies will be those that facilitate active decision-making, permitting the addict to choose not to take the drug. In contrast, transitioning from regulated relapse to social use or abstinence is best treated with a combination of pharmacology and behavioral interventions that reinforce and support correct decisions (Centonze et al, 2005). For example, behavioral interventions can range from classic approaches such as extinction training and cognitive behavioral therapy, to finding a steady job or reuniting with loved ones.
Stages of Addiction and Stages of Normal Reward Learning
Figure 1b attempts to map our current understanding of biological reward memory and learning processes onto the stages of addiction (Kelley, 2004; LaLumiere and Kalivas, 2006). Thus, the acquisition of memories and developing adaptive behavioral responses to important stimuli is referred to as acquisition and corresponds to social drug use. The counterpart to regulated relapse is the retrieval of declarative memories, that is, memories that are verbalized and are used in conscious decision-making. Finally, compulsive relapse can be considered equivalent to habit or procedural memories. The retrieval of procedural memories is not verbalized, and guides the unconscious execution of adaptive motor behaviors. These behaviors are well-learned and proceed most efficiently without ongoing decision-making (eg, riding a bike, or opening the refrigerator door when hungry).
There have been large advances over the last decade in our understanding of the underlying brain circuitry and neurotransmitters playing key roles in how motivational memories are acquired, and the learned behaviors executed. Interestingly, much of this knowledge has come through an iterative discovery process between researchers studying mechanisms of normal motivated learning and those studying drug-addiction as a pathology in normal learning. Figure 1b illustrates how key brain circuits and corresponding neurotransmitters map onto stages of addiction. Thus, learning to become addicted through social drug use critically involves dopamine cells in the ventral tegmental area (VTA) that release dopamine into the prefrontal cortex (PFC), amygdala, and nucleus accumbens (NA) (Berridge and Robinson, 1998; Kelley, 2004; Schultz, 2004; Wise, 2004; Jones and Bonci, 2005). One reasonable sequence supported by animal models is that as drug-seeking becomes well-learned, a reliance of the behavior on glutamatergic projections from PFC to the NA emerges (Pierce and Kalivas, 1997; Cardinal and Everitt, 2004; Wolf et al, 2004). Thus, regulated relapse strongly depends on the retrieval of drug-associated memories and the integration of these declarative memories through glutamatergic projections from the PFC to the NA. While glutamate continues to play a dominant role in compulsive relapse in this model, the glutamatergic circuit transitions from more declarative, executive prefrontal circuitry to habit circuitry involving classic cortico-striato-thalamic motor pattern generators, and the procedural memories that drive the unconscious engagement of well-learned behaviors (Barnes et al, 2005; Everitt and Robbins, 2005).
The remainder of this review involves a deeper dissection of the neuroplasticity underlying the stages of addiction, and an integration of this neuroplasticity into prospects for new drug development for transitioning addicts from compulsive to regulated relapse.
THE ACQUISITION OF ADDICTION THROUGH REPEATED DRUG EXPOSURE
As outlined in Figure 1, repeated drug intake (social drug use) involves the repeated release of dopamine from cells in the VTA into the PFC, striatal complex (including the NA), and amygdala. This circuit is illustrated in Figure 2a. Akin with motivationally relevant biological stimuli, all addictive drugs increase dopamine release within this circuit, albeit by different molecular mechanisms of action (Jay, 2003; Kelley, 2004; Nestler, 2005). This association between increased dopamine transmission and learning behaviors to obtain reward has led to an understanding that dopamine release is a key event to facilitate learning. Thus, many studies show that inhibiting dopamine transmission decreases motivation and learning, whereas stimulating dopamine typically promotes the acquisition of learned behaviors. The release of dopamine has been purported to imbue an event with salience, creating an internal sense that this is a relatively important event requiring the development of a behavioral response (Berridge and Robinson, 1998). Important distinctions exist between dopamine released by addictive drugs vs motivationally relevant environmental stimuli, and these distinctions are thought to be critical for the development of regulated and compulsive drug-seeking.

Corticolimbic dopaminergic pathways and a hypothetical comparison between dopamine release induced by motivationally important biological stimuli and addictive drugs. (a) Corticolimbic circuit regulating drug-seeking, including dopaminergic innervation from the VTA, to the PFC, basolateral amygdala (BLA), and NA; glutamatergic projections from the PFC and BLA to the nucleus accumbens; GABAergic/peptidergic projection from the NA to the VP. (b) Based primarily on the microdialysis literature, hypothetical changes in dopamine release in response to important biological stimuli (rewarding or aversive stimuli) vs the administration of addictive drugs. Note that drugs elicit more dopamine release for greater duration, and upon repeated administration (green arrows) tolerance does not develop to drug-induced dopamine release.
Figure 2b illustrates two major distinctions between dopamine release following motivational biological stimuli vs following exposure to an addictive drug. First, the release of dopamine by addictive drugs is of greater amplitude and duration than can be achieved through physiological mechanisms. Simply put, drug pharmacology drives dopamine release beyond physiological limits by overcoming normal homeostatic mechanisms for controlling dopamine release. For example, amphetamine-like psychostimulants inhibit the elimination of dopamine from synapses, and in some cases promote presynaptic dopamine release (Seiden et al, 1993), whereas other drugs such as nicotine or opioids act to alter feedback regulation of dopamine cells, causing an increase in dopamine cell activity. Thus, nicotine promotes excitatory glutamate transmission in the VTA, whereas opioids reduce the inhibitory GABA release on dopamine neurons (Nader and van der Kooy, 1997; Laviolette and van der Kooy, 2004; Pierce and Kumaresan, 2006). The second major difference shown in Figure 2b between drug-induced dopamine release and that produced by biological stimuli, is that tolerance develops to the release of dopamine by biological stimuli, whereas addictive drugs release dopamine every time the drug is taken. In chronic users, increased dosing is required due to tolerance, but with sufficient dose, a dopamine increase reliably occurs. An exception to this includes binges with amphetamine-like psychostimulants, which can cause short-term depletions of dopamine and chronic stimulant users who have reported extreme insensitivity or tolerance to the activating effects of the drug through as yet unknown mechanisms (Martinez et al, 2007). Thus, for biological rewards, once the person has learned the most efficient behavior to obtain a reward, dopamine release to facilitate further learning is not necessary and does not occur (Deutch and Roth, 1990; Schultz, 2004). However, it is important to note that dopamine continues to signal the arrival of a reward by conditioned stimuli (Schultz, 1998). For example, whereas delivery of food reward in response to a conditioned cue may no longer activate dopamine transmission in a trained animal, the appearance of a cue previously associated with food delivery will increase dopamine cell firing, presumably preparing the animal to initiate the adaptive food-seeking response. Thus, within physiological parameters, dopamine serves two functions, (1) to facilitate initial learning of adaptive responding to important stimuli, and (2) to cue the retrieval of the information needed to execute the adaptive behavioral response when environmental circumstances predict that food is imminent. In contrast, every administration of an addictive drug reward is associated with a large release of dopamine that can be expected to promote new learning (ie, new associations between the drug and environment) or reinforce prior learning, as well as to cue the addict to execute a drug-seeking behavior (ie relapse). In animal models, cues may also augment the response to stimulants thus producing a sensitized response to a given dose of the stimulant. In this way, repeated use of addictive drugs promotes increasing associations between the drug and life events, whereas biologically important stimuli do not. This may account for why repeated use of a drug causes drug-seeking behaviors to encroach upon all facets of daily life as the person becomes more dependent.
As indicated above, different drugs of abuse release dopamine via different molecular mechanisms. One of the actions of ethanol is activation of the endogenous opioid system so that if opiate receptors are blocked by an antagonist such as naltrexone, the alcohol-induced dopamine increase does not occur and the reward is blocked (Gonzales and Weiss, 1998). Thus, the behavioral manifestations of plasticity in human addicts may differ according to the drug. In heroin addicts, for example, repeated drug use produces marked tolerance with conditioned cues producing drug-opposite or withdrawal-like responses (O'Brien, 1975; O'Brien et al, 1977). Drug cues in cocaine addicts produce cocaine craving and limbic activation (Childress et al, 1999) with associated conditioned dopamine release (Volkow et al, 2006). Overall, in human addicts, tolerance is the neuroadaptation most often observed even in cocaine addicts (O'Brien et al, 2006). This results in increasing doses of self-administered drugs to achieve the drug effects originally obtained.
Dopamine-Induced Neuroplasticity Underlying Development of Regulated and Compulsive Relapse
The D1 and delta-FosB signaling cascade
The release of dopamine by important stimuli or addictive drugs produces changes in how neurons integrate excitatory and inhibitory neurotransmission. The effects of dopamine receptor activation are complex and distinctions exist between activation of D1-like vs D2-like receptors depending on the presence of pre- and postsynaptic localizations within the local circuitry of a given nucleus. There are a number of excellent reviews describing the current state of knowledge regarding dopamine signaling as it pertains to addiction and motivated learning (Berke and Hyman, 2000; Nicola et al, 2000; El-Ghundi, 2007). For our purposes, Figure 3 illustrates some key events precipitated directly by D1 receptor activation that are thought to be important antecedents for developing the enduring changes in neuronal physiology that underlie the establishment of adaptive behaviors to motivationally relevant events, as well as maladaptive drug-seeking behaviors. Importantly, this signaling cascade involves changes in gene transcription and chromatin remodeling which are thought to underlie the transition from social use to regulated and compulsive relapse. Thus, stimulation of D1 receptors in the striatum and cortex increases cAMP, cAMP-dependent protein kinase (PKA), and cAMP response element-binding protein (CREB) that promotes the transcription of many genes implicated in addiction, such as cfos, deltaFosB, Homer, and preprodynorphin (Hurd and Herkenham, 1993; Nestler et al, 2001; McClung and Nestler, 2003; Benavides and Bibb, 2004). Importantly, the rise of CREB in the NA and, to a lesser extent, the VTA has been linked to reduced drug-induced reinforcement (Carlezon et al, 1998; Nestler, 2005). Although not all addictive drugs appear to increase CREB in the accumbens (Pandey et al, 2004), overexpression of CREB in the accumbens inhibits the rewarding effects of psychostimulants, mu opioids, and biological rewards, whereas overexpression of a dominant-negative CREB mutant promotes drug reward (Barrot et al, 2002; Lu et al, 2003; McClung and Nestler, 2003). Interestingly, some studies show that CREB is necessary for the rewarding effects of addictive drugs and biological reinforcement (Jin et al, 2005; Walters et al, 2005; Choi et al, 2006), posing the possibility that whereas acute regulation of CREB is required for motivated behaviors, repeated upregulation of CREB induces tolerance to the reinforcing effects of rewarding stimuli. Certain CREB-regulated genes, such as preprodynorphin, NAC-1, and Homer, undoubtedly contribute to the compensatory effect increasing CREB to reduce the value of drug reward. For example, increased dynorphin inhibits the activity of dopamine cells and presynaptic dopamine release (Carlezon et al, 1998; Chefer et al, 2000; Hyman et al, 2006), and viral overexpression of NAC-1 or Homer1c in the accumbens inhibits the development of sensitized motor behaviors by repeated cocaine (Mackler et al, 2000; Szumlinski et al, 2006). Importantly, two of these proteins, preprodynorphin and NAC-1 show an enduring upregulation in abstinence, indicating a long-lasting compensatory inhibition of drug reward (Hurd and Herkenham, 1993; Cha et al, 1997). Unfortunately, as discussed in detail below, the devaluing of drug reward may also extend to biological rewards.

Dopamine D1 receptor-dependent signaling in spiny cells of the nucleus accumbens hypothesized to underlie the transition from social use to enduring vulnerability to relapse. By stimulating cAMP synthesis and ultimately phosphoryating and activating the transcriptional regulator CREB, a cascade of changes in protein synthesis occurs through the induction of additional transcriptional regulators (eg, c-Fos and ΔFosB). Also, the synthesis of proteins is induced that are important compensatory regulators of cellular functions and contribute to long-term drug-induced neuroplasticity (see text for discussions of specific proteins).
Of the CREB-regulated genes, the increase in the transcriptional regulator, deltaFosB, has proven particularly interesting (Nestler et al, 2001). The increase in many transcriptional regulators and immediate early genes by addictive drugs or biological motivational stimuli, such as cfos, Arc, Homer1a, and narp, diminishes after repeated exposure. In contrast, deltaFosB accumulates in dopamine-terminal fields in the cortex and striatum (Nestler et al, 2001; McClung and Nestler, 2003). This accumulation occurs in response to chronic administration of all drugs of abuse tested to date, as well as in response to repeated biologically motivating stimuli. Thus, the accumulation of deltaFosB is likely critical for learning and developing motivated behaviors in general. In the case of addictive drugs, pharmacological or genetic disruption of this cascade inhibits the development of certain forms of addiction-associated behavioral plasticity, such as sensitized motor behaviors (Nestler et al, 2001; McClung and Nestler, 2003). Akin to genes regulated by CREB, some of the genes directly regulated by deltaFosB may be compensatory and serve to limit drug reinforcement, and perhaps drug-seeking (Nestler, 2005). Thus, the induction of Cdk5 phsophorylates the dopamine-regulated phosphatase DARPP-32, thereby preventing its phosphorylation and activation by PKA (Benavides and Bibb, 2004). However, the induction of other genes by deltaFosB likely promotes drug reward and the majority of studies indicates that overexpression of deltaFosB increases drug reward (Kelz et al, 1999; Colby et al, 2003; Zachariou et al, 2006). Examples of deltaFosB gene regulation that would promote drug reward include the induction of GluR2 in the shell of the accumbens (Todtenkopf et al, 2006), and suppression of dynorphin expression (Zachariou et al, 2006). Importantly, the induction of deltaFosB and the gene products it regulates appears to be relatively transient and normalizes during abstinence. Therefore, although important for the acquisition of drug-seeking behaviors, deltaFosB itself is not an example of stable drug-induced neuroplasticity directly mediating the execution of regulated or compulsive relapse. Indeed, it is the transient nature of deltaFosB expression that makes it an ideal candidate for a protein mediating the transition from social use to relapsing drug use (Nestler et al, 2001). Accordingly, whereas deltaFosB regulated gene expression itself is transient, neuroplasticity regulated by these genes may be extremely stable during abstinence. For example, an enduring increase in dendritic spine density has been reported in accumbens spiny cells during extended abstinence from chronic psychostimulant administration (Robinson and Kolb, 2004), and this increase is mediated in part by deltaFosB stimulation of Cdk5 (Norrholm et al, 2003).
In summary, activation of the D1, CREB, and deltaFosB signaling cascade is clearly necessary for driving the neuroplasticity underlying both biological motivated learning and developing drug-seeking behaviors (Nestler, 2001; Hyman et al, 2006). However, the roles for drug-induced adaptations in this cascade in the execution of drug-seeking or in the vulnerability to relapse is complex. For example, the short-term and enduring neuroplasticity induced by activation of CREB is most often shown to serve a compensatory function to reduce dopamine or glutamate transmission in the accumbens, whereas increased deltaFosB regulates gene expression in a manner that is both compensatory (increased Cdk5) and supportive of drug reward (increased GluR2; decreased dynorphin). These adaptations in general would reduce the relative value of motivational biological stimuli, and this could indirectly contribute to the enduring vulnerability to relapse to drug-seeking. Thus, by functioning in a compensatory fashion to devalue all rewards, the enduring molecular consequences of transitory potentiation of the D1-CREB signaling cascade (eg, increased dynorphin, NAC1, and Homer1c) are promoting drug-seeking in favor of obtaining biological rewards.
Brain-Derived Neurotrophic Factor Regulation of Synaptic Plasticity in Addiction
Another dopamine-dependent change in protein synthesis that appears particularly important in establishing physiological as well as drug-induced neuroplasticity is a rise in brain-derived neurotrophic factor (BDNF). BDNF is in the class of psychostimulant-regulated immediate early genes, including Arc, c-fos, and zif/268 (Dunais and McGinty, 1994; Moratalla et al, 1996). However, BDNF and Arc appear unique because their mRNA is strongly induced and transported into dendrites by cellular activity (Steward and Worley, 2001). Of particular interest, and apparently distinct from genes regulated by deltaFosB, as well as other activity-dependent genes upregulated by psychostimulants, the enduring changes in BDNF accumulate with increasing periods of abstinence (Grimm et al, 2003; Lu et al, 2004a; Filip et al, 2006). Also, stimulating BDNF receptors in the amygdala, NA, or VTA promotes (Horger et al, 1999; Lu et al, 2004b; Graham et al, 2007; Pu et al, 2006), whereas microinjection of BDNF into the PFC inhibits drug-seeking (Berglind et al, 2007), indicating that akin to activation of deltaFosB, BDNF serves a general physiological role in supporting neuroplasticity that is usurped by addictive drugs to ultimately establish regulated and compulsive relapse.
BDNF is well known to promote forms of excitatory synaptic plasticity, such as early- and late-phase long-term potentiation (LTP), and also promote dendritic spine formation (Bramham and Messaoudi, 2005). The mechanisms underlying what is in general an enhancement in excitatory transmission are varied, and include increasing synaptic vesicle docking, increasing glutamate release, and promoting postsynaptic NMDA signaling. Given these cellular mechanisms, it is not surprising that BDNF has been implicated in the neuroplasticity underlying normal learning and memory processes. Regarding drug addiction, BDNF mediates the enduring potentiation of excitatory transmission onto dopamine cells in the VTA elicited by repeated cocaine administration (Pu et al, 2006), and along with orexin release (Borgland et al, 2006), could contribute to the exciting series of observations of potentiated LTP in VTA dopamine cells following a single administration of an addictive drug (for review of these findings and how they may contribute to the induction of enduring forms of neuroplasticity that underlie relapse, see Jones and Bonci, 2005). Importantly, the level of BDNF in the VTA, as well as NA and amygdala, progressively increases during abstinence (Grimm et al, 2003). This progressive increase has been hypothesized to underlie the progressive increase in drug-seeking that occurs during cocaine withdrawal, which may occur, in part, by increasing dopamine D3 receptor expression (Guillin et al, 2001; Le Foll et al, 2005). The fact that BDNF is elevated by acute drug administration and also remains elevated in certain brain areas after extended abstinence marks this protein as a stable neuroplasticity candidate that may contribute to both the acquisition of drug-seeking, and the execution of drug-seeking after extended periods of abstinence.
Transitory Neuroplasticity Associated with the Molecular Site of Drug Action
Other relatively transitional forms of neuroplasticity induced by addictive drugs have also been described. However, in contrast to the D1-CREB-deltaFosB signaling pathway, these signaling events are more specific to individual drugs. For example, changes in dopamine transporters are associated with amphetamine-like psychostimulants (Daws et al, 2002), GABA-A receptor changes have been noted after chronic alcohol (Charlton et al, 1997), and nicotine desensitizes nicotinic receptors (Mansvelder and McGehee, 2000). These drug-specific changes contribute important nuances of addiction to each drug, in particular, withdrawal syndromes contain characteristics unique for each drug class. Also, drug-specific changes influence the circuitry critical for normal reward and drug learning. In general, the drug-specific effects are beyond the scope of the present review which is focused on what appear to be common features of brain plasticity shared by most or all drugs of abuse and in general principle, are also shared with motivational biological stimuli.
Summary of Neuroplasticity Underlying the Acquisition of Drug Use and the Transition from Social Drug Use to Regulated and Compulsive Relapse
Figure 4 illustrates different temporal categories of neuroplasticity associated with the repeated use of addictive drugs and subsequent abstinence. It is important to note that experiments conducted with repeated psychostimulant administration, and to a lesser extent opioids, provide the majority of information underlying the patterns shown in Figure 4a. Three general categories are proposed. The first category includes the induction of activity-dependent genes by acute administration, and the development of tolerance to this induction following repeated administration. Proteins in this category include c-fos, Arc, Homer1a, narp, and zif/268. Importantly, following a period of abstinence, tolerance subsides and these proteins can again be induced by acute psychostimulant treatment, often at levels or with patterns of expression different from that induced by the first drug exposure. These proteins are thought to be critical for initiating the neuroplasticity necessary to acquire new behaviors, as well as reconsolidate learned behaviors, including drug-seeking.

Stages of neuroplasticity in addiction. (a) Transient forms of neuroplasticity, typically involving the development of tolerance with repeated administration; potentially important in promoting social drug use. (b) Forms of plasticity that augment with repeated drug administration that diminish within hours to weeks after discontinuing drug administration; thought to be important in the transition from social to relapsing drug use. (c) Stable forms of plasticity emerging either during repeated drug use or during abstinence. In some instances, protein changes in this category progressively increase during abstinence, and are thought to contribute to the enduring vulnerability to relapse that is a cardinal feature drug addiction. Small arrows indicate repeated drug administration.
The second category is characterized by proteins whose expression gradually increases or decreases with repeated drug exposure, and endures for various periods of abstinence. Two subcategories are shown in Figure 4b. The first includes protein changes that endure for hours to days into abstinence and typically correspond to changes closely associated with the drug's molecular site of action. The other subcategory is typified by the accumulation of deltaFosB, where elevated levels can endure for days or weeks. This latter subcategory is thought to contribute to the acquisition of motivated learning, but importantly, in response to repeated drug use, deltaFosB has been hypothesized to mediate the transition for social drug use to relapsing use (Nestler, 2005).
The third category contains proteins that are elevated or reduced after prolonged abstinence. Two subcategories are considered in Figure 4c. The first is typified by BDNF that accumulates in certain brain regions after repeated psychostimulant administration and this accumulation progresses with increasing duration of abstinence (Grimm et al, 2003; Lu et al, 2004a). The second subcategory will be considered in more detail below, and contains proteins that do not change markedly during drug administration, but are elevated or reduced during abstinence. It is hypothesized that this category contains neuroplastic events that are likely responsible for the state of vulnerability to relapse. Most of these enduring changes have not been shown to occur in response to repeated exposure to motivational biological stimuli and may be biomarkers for the neuropathology of addiction.
ENDURING NEUROPLASTICITY THAT MAY PROMOTE THE VULNERABILITY TO RELAPSE
As described above, once a behavior designed to obtain a reward or avoid a negative consequence has been learned, the role for dopamine changes from one of promoting new learning to one of enabling the use of learned information to efficiently execute the adaptive behavioral response (Schultz, 2004). In contrast, glutamate transmission from cortex and allocortex (eg, amygdala and hippocampus) into the striatal motor circuit (including the NA) emerges as critical for executing a learned behavior (Kalivas and Volkow, 2005). Furthermore, it is thought that as a behavior is repeatedly executed, the role of corticofugal glutamate projecting from PFC and amygdala into the NA, becomes less important in favor of glutamate projecting from sensory motor cortical areas to the dorsal striatum (Everitt and Robbins, 2005). In this way, behavior evolves from being a declarative process involving prefrontal executive functions into a habitual behavior utilizing working memory circuitry (Barnes et al, 2005). Physiologically, this transition from declarative to automatic behaviors can be adaptive by permitting well-learned behaviors to proceed efficiently without conscious involvement, and if the motivationally important stimulus or context changes, executive functions intrude to disrupt the habit as part of developing a new adaptive behavior appropriate to the environmental change. In the case of drug-seeking, this transition from prefrontal circuitry to habit motor circuitry signifies loss of control and compulsive relapse. Important for the pathology of addiction, the capacity of prefrontal, declarative circuitry to intrude and disrupt the drug-seeking habit is impaired, making it more difficult for executive decision to intrude in drug-seeking (Everitt and Robbins, 2005; Kalivas and Volkow, 2005). Understanding the neurophysiology that reinforces this maladaptive transition from regulated to compulsive drug-seeking, and the impairment in reinvigorating prefrontal control over drug-seeking requires understanding the enduring cellular neuroplasticity wrought by repeated drug use. In particular, this involves identifying changes in glutamate transmission and the emergence of the hypofrontality that permits drug-seeking to proceed without conscious interventions (Jentsch and Taylor, 1999; Goldstein and Volkow, 2002).
Enduring Neuroplasticity in Cortical Glutamate Circuitry: Human Neuroimaging
Much of the neuroplasticity in cortical circuits has been visualized directly in addicts using various neuroimaging approaches. Thus, there is a general reduction in prefrontal cortical measures of cellular metabolism and blood flow in individuals addicted to a variety of different drugs, ranging from cocaine to opioids to alcohol (Goldstein and Volkow, 2002). This includes regions such as the anterior cingulate and ventral orbital cortex. Given the association between activation of the anterior cingulate and engaging biologically relevant motivated behaviors (Rilling et al, 2002), and between activation of the ventral orbital cortex and the ability to switch a well-learned behavior to a new adaptive behavior (Kolb et al, 2004), this hypofrontality has been characterized as a strong indicator of reduced ability to regulate drug-seeking. A reduction in frontal cortical neurons has also been reported in drug-free cocaine addicts (Franklin et al, 2002), but it is unknown whether these functional and anatomical indications of hypofrontality were vulnerability factors present before cocaine use or the effects of chronic stimulant use. Interestingly, when exposed to a cue previously associated with drug use that precipitates the desire for drug, there is marked activation in the PFC, including anterior cingulate and ventral orbital cortices (Goldstein and Volkow, 2002; Wilson et al, 2004; Kalivas and Volkow, 2005). In many studies, increased activity in the PFC has been positively correlated with the intensity of cue-induced desire for the drug. Thus, the differential in prefrontal activity between baseline and levels stimulated by drug cues is larger than what occurs in a control subject in response to cues associated with biological reward, such as a sexually evocative visual stimuli. Moreover, consistent with addiction being characterized in part by reduced response to biological rewards, when cocaine addicts were presented with a sexual stimulus, prefrontal activation was significantly impaired compared to controls (Garavan et al, 2000). Efforts to resist craving in response to cocaine cues have also been reported to increase frontal lobe activity (Childress et al, 2007), suggesting that impaired frontal function could be implicated in inability to resist relapse.
Another striking finding from neuroimaging studies indicating reduced response to biological reward in addicts is a reduction in dopamine receptor activation in response to low doses of psychostimulants (Volkow et al, 2004, 2005). Thus, methylphenidate-induced dopamine release into the striatum is impaired in cocaine addicts relative to control subjects. Also, regardless of the predominant drug being abused, addicts show reduced levels of D2 receptors in the striatum (Volkow et al, 2004). In as much as reduced D2 receptors indicates blunting of dopamine transmission, it is not surprising that addicts report reductions in high or pleasure in response to methylphenidate relative to control subjects. In contrast, whereas the methylphenidate induces strong cravings in the addicts, there is no craving in comparison subjects. However, non-drug using control subjects vary according to their striatal D2 receptor density. Those with low D2 density report positive, pleasurable effects from methylphenidate, whereas those with higher D2 density do not like the effects of the stimulant (Volkow et al, 2002). This finding in normal human subjects is paralleled by a similar finding in nonhuman primates (Nader and Czoty, 2005).
Taken together, these neuroimaging studies point to enduring changes in mesocorticolimbic circuitry. Thus, in the baseline state, the addict is relatively hyporesponsive to biological motivational stimuli, as evidenced by two neuroadaptations, (1) reduced activity in PFC, and (2) reduced striatal levels of dopamine D2 receptors. Perhaps even more critical to the pathology of addiction, the capacity for biologically relevant stimuli to activate PFC is impaired. Similarly, pharmacologically induced release of dopamine in the striatum and the corresponding subjective sensation of high or pleasure is impaired. However, drug-associated stimuli markedly activate PFC in addicts in a manner correlated with drug desire. Taken together, these neuroimaging data provide a neurocircuitry template for the prime features of addiction; excessive, uncontrolled responding for drug, and poor or inappropriate responding for biologically important stimuli.
Enduring Neuroplasticity in Cortical Glutamate Circuitry: Animal Models
To understand the cellular basis for how these changes in corticolimbic circuitry occur and hopefully identify mechanisms for reversing or countering the changes, it is necessary to use animal models, which permit a more mechanistic analysis. Importantly, animals self-administer drugs that are addictive in humans, and the circuitry-based transition from the dopamine-dependent acquisition of drug-seeking to the glutamate-dependent execution of drug-seeking is evident in animals studies.
The most widely employed model of relapse involves training rats to self-administer a drug, placing the animal in forced abstinence either with or without extinction training, then re-exposing the animals to the drug context, cues specifically paired with drug delivery, stress or the drug itself (Epstein et al, 2006). In response to these stimuli, the drug-trained animal will engage in drug-seeking even in the absence of obtaining drug.
Early studies of relapse in animals involved the use of opiate-dependent rats treated with naloxone or naltrexone. Opiate reward was blocked and after initial increased responding, rapid decrements occurred (Davis and Smith, 1974). More recently, alcohol self-administration was found to activate the endogenous opioid system producing dopamine release in the NA and alcohol seeking in the animal (Gonzales and Weiss, 1998). Rats trained to self-administer alcohol and then given naltrexone will show a cessation of dopamine increase in the NA and cessation of alcohol self-administration. This straightforward extinction model is mirrored in human alcoholics who report decreased or absent alcohol reward when treated with naltrexone (Volpicelli et al, 1995).
More recently, inactivation of various brain nuclei with GABA agonists or compounds inhibiting action potentials, the brain nuclei necessary to execute drug-seeking have been mapped (McFarland and Kalivas, 2001; See, 2002; McFarland et al, 2004). The results of these studies conducted over the last decade are remarkably parallel with the afore-mentioned human imaging studies. The dorsolateral striatum is the brain region that has been shown to be obligatory regardless of the modality for inducing drug-seeking, or the presence or absence of extinction training. This identifies the involvement of habit motor circuitry in a well-trained behavior such as drug-seeking. Surprisingly, unless animals undergo extinction training, the drug-seeking induced by placing the animals back in the drug context is not affected by inhibiting any other brain structure classically associated with motivated learning or cue-induced craving in neuroimaging studies of addicts (eg, areas of PFC, amygdala, or NA) (Fuchs et al, 2006). However, if the animal undergoes extinction training, drug-seeking induced by cues, stress, or the drug itself engages a much more enriched circuit, containing the circuitry identified in human imaging of addicts. For example, if an experimenter locally inhibits any of the nuclei in the series circuit containing the dopamine projection from the VTA to the dorsal PFC, the glutamate projection from the PFC to the NA or the GABA/peptide projection from the accumbens to the ventral pallidum (VP), drug-seeking in an extinguished animal is blocked. Thus, extinction training engages brain regions involved in more declarative and emotional processing in drug-seeking behavior (McFarland and Kalivas, 2001; See, 2002; McFarland et al, 2004), implying the assertion of executive behavioral modulation. Corresponding to extinction training bringing behavioral control, the amount of drug-seeking (eg, lever pressing) induced by the drug context in abstinent animals is higher than drug-seeking induced in extinguished animals (Fuchs et al, 2006). Together, the circuitry and behavioral data indicate that the more enriched circuit associated with drug-seeking in extinguished subjects serves to regulate drug-seeking. Supporting this possibility, extinction training induces GluR1 and GluR2 glutamate receptor subunits in the NA of cocaine-trained rats (Sutton et al, 2003). Similarly, extinction training in fear-conditioned animals involves activation of the infralimbic cortex that projects to the NA (Sierra-Mercado et al, 2006). Thus, just as psychosocial interventions in human addicts endeavor to restore executive control over drug-seeking habits, extinction training in animals engages a more enriched prefrontal circuit that modulates the drug-seeking in response to cues, stress, or the drug itself.
The similarities in prefrontal circuitry between drug-trained animals and human addicts is further reflected by a dramatic elevation in glutamate transmission during extinguished drug-seeking. Thus, rats trained to self-administer cocaine or heroin show a marked increase in synaptic glutamate release in the NA in response to drug- or stress-induced drug-seeking (McFarland et al, 2003, 2004). Moreover, this rise is abolished by inhibition of the dorsal PFC, and does not occur in either yoked saline or yoked cocaine or heroin control groups. In other words, regardless of repeated drug administration, if the animals do not engage in drug-seeking behavior there is not dysregulated release of synaptic glutamate. Accordingly, acute drug administration alone is not sufficient to activate the prefrontal accumbens glutamate pathway, rather this pathway is recruited by animals learning a drug-seeking task. Importantly, no increase in glutamate was observed during food-seeking in animals trained to self-administer food, indicating that this neuroplasticity is not induced by learning to seek biological rewards (McFarland et al, 2003). Supporting the importance of the dysregulation in glutamate release in driving drug-seeking behavior, intra-accumbens administration of glutamate antagonists prevents drug seeking, as does inactivation of the PFC (Cornish and Kalivas, 2000; Di Ciano and Everitt, 2001). Recently, some of the molecular neuroplasticity mediating the dysregulation of the prefrontal glutamate projection to the NA has been studied. In addition, some enduring consequences of repeated release of glutamate during drug-seeking have been investigated.
Neuroplasticity Contributing to Dysregulated Glutamate Transmission
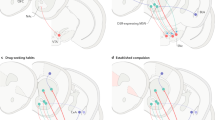
As the augmented release of glutamate is enduring, the corresponding molecular plasticity is also enduring. Key among these molecular adaptations is downregulation of cystine–glutamate exchange (xc−) (Baker et al, 2003). xc− is the rate-limiting step whereby cells acquire cystine to make the intracellular antioxidant glutathione, and occurs by exchanging the uptake of one cystine in exchange for the release of one molecule of intracellular glutamate into the extracellular space (McBean, 2002). Normally, this nonsynaptic glutamate release results in levels in the extracellular space sufficient to stimulate inhibitory presynaptic metabotropic glutamate autoreceptors (mGluR), and thereby dampen synaptic glutamate release (Moran et al, 2005). However, reduced xc− in the NA after chronic cocaine removes this tonic inhibition, increasing synaptic glutamate release probability. This reduction in tone is combined with reduced signaling through the presynaptic mGluRs, that is thought to result from increased receptor phosphorylation (Xi et al, 2002), and the induction of a protein called activator of G-protein signaling 3 (AGS3), which serves to limit receptor signaling through the Giα class of G proteins (Blumer and Lanier, 2003; Bowers et al, 2004; Yao et al, 2005). This relationship is illustrated in Figure 5.

Molecular neuroplasticity associated with excitatory synapses in the NA hypothesized to underlie the vulnerability to relapse to cocaine and perhaps other addictive drugs. Stimuli eliciting a learned behavior to approach a natural award result in glutamate release in the prefrontal to accumbens pathway that is well regulated. A stimulus eliciting cocaine-seeking results in massive release of glutamate that can be measured as overflow in the extracellular fluid. Dysregulated release results in part from downregulated xc− and reduced activation of inhibitory mGluR presynaptic receptors. The repeated massive release of glutamate also promotes dendritic dysmorphisms, including increased spine density as a result of increased actin cycling. Increasing intensity of the red coloring in the extracellular space denotes increasing concentration of glutamate, and the green circles correspond to cystine.
The repeated dysregulated release of synaptic glutamate during repeated drug-seeking episodes is thought to contribute to a number of enduring postsynaptic changes. Primary among these is the well-established changes in dendritic spine density seen in the NA and prefrontal cortical areas after repeated administration of addictive drugs (Robinson and Kolb, 2004). It is well established that applying glutamate to neurons in culture changes spine density, either increasing or decreasing depending on the amount of glutamate receptor stimulation and perhaps the subtypes stimulated (Lippman and Dunaevsky, 2005; Richards et al, 2005). Thus, it is perhaps not surprising that depending on which drug is chronically administered, there is an increase (psychostimulants) or decrease (opioids) in spine density (Robinson and Kolb, 1999, 2004; Jedynak et al, 2007). The underlying cellular mechanisms of neuroplasticity regulating spine morphology is an emerging area of extremely intense research activity. However, the regulation of the actin cytoskeleton that can both stabilize or change spine morphology is a prime candidate for a process that may underlie changes in spine density (Rao and Craig, 2000; Lisman, 2003; Blanpied and Ehlers, 2004; Matus, 2005). Accordingly, there is an enduring increase in actin cycling after withdrawal from chronic psychostimulant administration (Toda et al, 2006). The increase in actin cycling occurs, at least in part, from a reduction in Lim kinase, which critically regulates F-actin depolymerization, as well as spine maturation (Meng et al, 2002; Soosairajah et al, 2005). In addition to alterations in spine morphology another consequence of increased actin cycling would be alterations in the trafficking of proteins into the postsynaptic membrane (Kasai et al, 2003). Although not necessarily the result of increased actin cycling, a potentially critical change in postsynaptic receptor trafficking is an enduring increase in membrane insertion of AMPA glutamate receptors (Mangiavacchi and Wolf, 2004; Sun et al, 2005; Boudreau and Wolf, 2005). Surprisingly, however, the increase in AMPA receptors is associated with an inability to induce long-term depression (which is generally associated with reduced AMPA receptors) (Martin et al, 2006). Although this finding has recently been contested in a study showing that after withdrawal from cocaine there is a marked increase in AMPA current in accumbens spiny cells (Kourrich et al, 2007). In general, the electrophysiological correlates of addiction in accumbens spiny cells is currently an area of some confusion in the literature (Kalivas and Hu, 2006).
Interestingly, stimulating BDNF receptors promotes actin cycling and modulates spine density (Bramham and Messaoudi, 2005), indicating that the afore-mentioned progressive elevation of BDNF during withdrawal may contribute directly to the enduring adaptations in excitatory transmission. In apparent contradiction to this hypothesis, stimulating BDNF receptors in the accumbens promotes cocaine-seeking (Graham et al, 2007), an effect also elicited by inhibiting actin cycling in the NA (Toda et al, 2006). However, a recent study revealed that the release of BDNF into the accumbens after administration into the PFC prevented both cocaine-induced drug seeking and the release of glutamate associated with cocaine-seeking (Berglind et al, 2007). It was speculated that the BDNF administered into the PFC was anterogradely transported and release into the NA to produce this behavioral effect (Altar et al, 1997). Thus, the endogenous release of BDNF from prefrontal afferents into the NA may produce a different effect than microinjecting pharmacological quantities.
Although enduring neuroplasticity in the NA and striatum may reflect the hypofrontality observed in neuroimaging addicts, it is assumed that enduring neuroplasticity is also occurring directly in the PFC. Indeed, repeated psychostimulant administration increases dendritic spine density on prefrontal pyramidal cells (Robinson and Kolb, 2004). In contrast to spiny cells in the accumbens where increased spine density is associated with less intrinsic membrane excitability (Zhang et al, 1998), prefrontal pyramidal cells appear to be more easily stimulated (Dong et al, 2005). This is commensurate with the large increase in synaptically released glutamate in the NA produced during drug-seeking, and may, in part, be related to cellular neuroadaptations such as reduced signaling through Gi-coupled receptors due to elevated AGS3 (Kalivas et al, 2005). Accordingly, while D2 receptor-mediated changes in prefrontal cell firing appear blunted after withdrawal from chronic cocaine, the effects of activating Gs-coupled D1 receptors are augmented (Nogueira et al, 2006). This may contribute to the increased excitability and loss of membrane bistability reported in prefrontal neurons after chronic cocaine (Trantham et al, 2002), as D1 receptor stimulation promotes AMPA receptor insertion into the membrane (Sun et al, 2005). The fact that D1 receptor stimulation in the PFC is required for reinstating drug-seeking is consistent with this possibility (Capriles et al, 2003; Sun and Rebec, 2005).
Summary of Neuroplasticity Underlying the Execution of Regulated and Compulsive Relapse
As shown in Figure 4c, forms of neuroplasticity that endure during abstinence provide the neuroplastic substrates underlying the enduring vulnerability to relapse in addiction. A variety of studies support increased prefrontal glutamate release into the NA as a critical mediator of drug-seeking. Similarly, marked alterations in postsynaptic glutamate signaling, including morphological changes in striatal neurons likely contribute to the changes. The cellular plasticity underlying both the hypofrontality apparent during baseline and the strong response of PFC and outputs to the NA during drug-seeking or drug desire is beginning to be elucidated and, as outlined below, constitute new potential sites of action for developing pharmacotherapies for treating addiction.
FUTURE DIRECTIONS AND CLINICAL IMPLICATIONS
As we increase our understanding of the circuitry and cellular mechanisms by which repeated drug exposure increases the vulnerability to relapse, new potential drug targets become apparent. How this vulnerability transitions between regulated and compulsive relapse affords rationales for the development of new medications, as well as increased understanding of ways that medications might improve the results of psychosocial therapies.
Converting Compulsive to Regulated Relapse
Using pharmacotherapies to facilitate the capacity of addicts to involve more declarative, decision-making processes in relapse is critical to decreasing compulsive relapse. As outlined above, the transition to relapse becoming a habit based on unconscious working memory circuitry involves a loss of prefrontal regulation. With some drugs of abuse this is manifested by cognitive deficits in functions related to attention, impulsivity and the ability to alter behavior based on new information. Based on these findings, pharmacologically normalizing or countering neuroplasticity produced in the PFC to regulate striatal habit circuitry would seem a valuable approach. As outlined above, the molecular changes involve an apparent downgrading of biological reward through reduced dopamine transmission, and augmented prefrontal to accumbens glutamate transmission to drive forward drug-seeking. Thus, drugs that alter dopamine transmission, glutamate transmission or GABA transmission are potential candidates. Moreover, the GABA projection from the NA is colocalized with a variety of neuropeptides (McGinty, 2007), and these peptides, as well as others in corticolimibic are also candidates for drug development.
Dopaminergics
Dopamine transmission undergoes differential changes depending on the receptor subtype. Thus, there is a reduction in D2 receptor signaling (Volkow et al, 2004), a potential increase in D1 signaling (Kalivas et al, 2005), and a marked elevation in D3 receptors as a result of enduring increases in BDNF (Neisewander et al, 2004). This makes it difficult to predict how to best target dopamine transmission. However, there is excellent preclinical data supporting use of D3 antagonists to inhibit drug-seeking (Xi et al, 2006).
Glutamatergics
Based on the neuroplasticity outlined above, blocking the release of synaptic glutamate associated with drug-seeking would seem to be an excellent approach to diminish the motivation to relapse. However, it is not possible to use full antagonists of ionotropic glutamate receptors due to unacceptable side effects. Accordingly, a variety of pharmacological mechanisms are emerging for modulating rather than blocking glutamate transmission. Some of these compounds have already entered clinical trials and show modest efficacy. For example, acamprosate and topiramate have weak actions as AMPA receptor antagonists (Myrick and Anton, 2004; Cubells, 2006). Topiramate has been reported to reduce relapse in cocaine addicts (Kampman et al, 2004). Also, modafinil and N-acetylcysteine acting to increase extracellular glutamate and thereby stimulating mGluR-induced inhibition of synaptic glutamate release have shown efficacy in cocaine relapse or cue-induced craving, respectively (Dackis et al, 2005; LaRowe et al, 2007). Three independent labs have reported (Dackis, 2004; Malcolm et al, 2006; Hart et al, 2007) that modafinil reduces the cocaine high possibly by increasing extracellular glutamate and activating inhibitory mGluR as described above. Moreover, in preclinical models, mGluR2/3 agonists have been shown to inhibit drug-seeking (Baptista et al, 2004; Peters and Kalivas, 2006).
GABAergics
Preclinical models of cocaine and heroin show that decreased GABA release into the VP by NA afferents is associated with drug-seeking (Caille and Parsons, 2004; Tang et al, 2005). Supporting the importance of this adaptation, drugs that promote GABA transmission have shown promise in preclinical and clinical studies, vigabatrin (inhibitor of GABA transferase), gabapentin (mechanism unclear), and baclofen (GABAb agonist). The reader is referred to recent overviews of the use of GABAergics in treating drug addiction (O'Brien, 2005; Vocci and Ling, 2005).
Peptidergics
Many neuropeptides are colocalized with GABA in the projection from the NA, including neurotensin, substance P, dynorphin, and CART (McGinty, 2007). Although our knowledge of how these peptides contribute or not to regulating drug-seeking mediated by the accumbens to pallidum projection is relatively poor, it has been shown that blocking enkephalin receptors in the VP prevents cocaine-seeking in animal models (Tang et al, 2005), an effect perhaps contributing the utility of naltrexone in ethanol addiction (Vocci and Ling, 2005).
Conclusions
Although there have been important advances in our understanding of the neuroplasticity underlying the development of addiction and the enduring vulnerability to relapse, we are at a very nascent stage in being able to apply this new knowledge to treat addicts. Although we have some established pharmacological candidates for regulating neurotransmission between neurons in the circuitry shown to be important, it is difficult to manipulate the neuroplasticity produced in intracellular signaling that is so critical to addiction. Using this new information awaits the development of compounds selective for targeted proteins in the signaling pathways, and more importantly, means to deliver the compounds. Nonetheless, the forms of neuroplasticity identified to date point the way towards future therapies that will become available as the delivery technology evolves.
References
Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL et al (1997). Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 389: 856–860.
APA (1987). Diagnostic and Statistical Manual of Mental Disorders. Third Edition, Revised (DSM III-R). In: American Psychiatric Association: Washington, DC.
Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S et al (2003). Neuroadaptations in cystine–glutamate exchange underlie cocaine relapse. Nat Neurosci 6: 743–749.
Baptista MA, Martin-Fardon R, Weiss F (2004). Preferential effects of the metabotropic glutamate 2/3 receptor agonist LY379268 on conditioned reinstatement vs primary reinforcement: comparison between cocaine and a potent conventional reinforcer. J Neurosci 24: 4723–4727.
Barnes TD, Kubota Y, Hu D, Jin DZ, Graybiel AM (2005). Activity of striatal neurons reflects dynamic encoding and recoding of procedural memories. Nature 437: 1158–1161.
Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ et al (2002). CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci USA 99: 11435–11440.
Benavides DR, Bibb JA (2004). Role of Cdk5 in drug abuse and plasticity. Ann N Y Acad Sci 1025: 335–344.
Berglind W, See R, Fuchs R, Branham R, Whitfield T, Miller S et al (2007). A BDNF infusion into the medial prefrontal cortex suppresses cocaine-seeking behavior. Eur J Neurosci 26: 757–766. Demonstration that transynaptic release of BDNF can ameliorate drug-seeking.
Berke JD, Hyman SE (2000). Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25: 515–532.
Berridge K, Robinson T (1998). What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Rev 28: 309–369.
Blanpied TA, Ehlers MD (2004). Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry 55: 1121–1127.
Blumer J, Lanier SM (2003). Accessory proteins for G protein-signaling systems: activators of G protein signaling and other nonreceptor proteins influencing the activation state of G proteins. Receptors Channels 9: 195–204.
Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A (2006). Orexin A in the VTA is critical for the induction o f synaptic plasticity and behavioral sensitization to cocaine. Neuron 49: 589–601.
Boudreau AC, Wolf ME (2005). Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci 25: 9144–9151.
Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML et al (2004). Activator of G-protein signaling 3: a gatekeeper of cocaine sensitization and drug-seeking. Neuron 42: 269–281.
Bramham CR, Messaoudi E (2005). BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol 76: 99–125.
Caille S, Parsons LH (2004). Intravenous heroin self-administration decreases GABA efflux in the ventral pallidum: an in vivo microdialysis study in rats. Eur J Neurosci 20: 593–596.
Capriles N, Rodaros D, Sorge RE, Stewart J (2003). A role for the prefrontal cortex in stress- and cocaine-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 168: 66–74.
Cardinal RN, Everitt BJ (2004). Neural and psychological mechanisms underlying appetitive learning: links to drug addiction. Curr Opin Neurobiol 14: 156–162.
Carlezon WA, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N et al (1998). Regulation of cocaine reward by CREB. Science 282: 2272–2274.
Centonze D, Siracusano A, Calabresi P, Bernardi G (2005). Removing pathogenic memories: a neurobiology of psychotherapy. Mol Neurobiol 32: 123–132.
Cha X-Y, Pierce RC, Kalivas PW, Mackler SA (1997). NAC-1, a rat brain mRNA, is increased in the nucleus accumbens three weeks after chronic cocaine self-administration. J Neurosci 17: 6864–6871.
Charlton ME, Sweetnam PM, Fitzgerald LW, Terwilliger RZ, Nestler EJ, Duman RS (1997). Chronic ethanol administration regulates the expression of GABAA receptor α1 and α5 subunits in the ventral tegmental area and hippocampus. J Neurochem 68: 121–127.
Chefer VI, Moron JA, Hope B, Rea W, Shippenberg TS (2000). Kappa-opioid receptor activation prevents alterations in mesocortical dopamine neurotransmission that occur during abstinence from cocaine. Neuroscience 101: 619–627.
Childress AR, Mozley PD, McElgin W, Fitzgeral J, Reivich M, O'Brien CP (1999). Limbic activationduring cue-induced cocaine craving. Am J Psychiatry 156: 11–18.
Childress AR, Wang Z, Li Z, Erman R, Hole A, MacDouball M et al (2007). Brain substrates for cue-induced cocaine craving (GO!) and it sinhibition (STOP) as revealed by machine classifier learning. College on Problems of Drug Dependence Annual Meeting, Quebec City (abstract).
Choi KH, Whisler K, Graham DL, Self DW (2006). Antisense-induced reduction in nucleus accumbens cyclic AMP response element binding protein attenuates cocaine reinforcement. Neuroscience 137: 373–383.
Colby CR, Whisler K, Steffen C, Nestler EJ, Self DW (2003). Striatal celltype-specific overexpression of DeltaFosB enhances incentive for cocaine. J Neuroscience 23: 2488–2493.
Cornish J, Kalivas P (2000). Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci 20 (RC89): 81–85.
Cubells JF (2006). Topiramate for cocaine dependence. Curr Psychiatry Rep 8: 130–131.
Dackis CA (2004). Recent advances in the pharmacotherapy of cocaine dependence. Curr Psychiatry Rep 6: 323–331.
Dackis CA, Kampman KM, Lynch KG, Pettinati HM, O'Brien CP (2005). A double-blind, placebo-controlled trial of modafinil for cocaine dependence. Neuropsychopharmacology 30: 205–211.
Davis WM, Smith SG (1974). Naloxone use to eliminate opiate-seeking behavior: need for extinction of conditioned reinforcement. Biol Psychiatry 9: 181–189.
Daws LC, Callaghan PD, Moron JA, Kahlig KM, Shippenberg TS, Javitch JA et al (2002). Cocaine increases dopamine uptake and cell surface expression of dopamine transporters. Biochem Biophys Res Commun 290: 1545–1550.
Deutch AY, Roth RH (1990). The determinants of stress-induced activation of the prefrontal cortical dopamine system. Prog Brain Res 85: 357–393.
Di Ciano P, Everitt BJ (2001). Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology 25: 341–360.
Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT et al (2005). Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci 25: 936–940.
Dunais JB, McGinty JF (1994). Acute and chronic cocaine administration differentially alters striatal opioid and nuclear transcription factor mRNAs. Synapse 18: 35–45.
El-Ghundi M, O'Dowd BF, George SR (2007). Insights into the role of dopamine in learning and memory. Rev Neurosci (in press).
Epstein DH, Preston KL, Stewart J, Shaham Y (2006). Toward a model of drug relapse: an assessment of the validity of the reinstatement procedure. Psychopharmacology (Berl) 189: 1–16.
Everitt BJ, Robbins TW (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 8: 1481–1489. First clear articulation of the transition from prefrontal glutamate to cortico-striatal glutamate as the addict transitions from regulated to compulsive relapse.
Filip M, Faron-Gorecka A, Kusmider M, Golda A, Frankowska M, Dziedzicka-Wasylewska M (2006). Alterations in BDNF and trkB mRNAs following acute or sensitizing cocaine treatments and withdrawal. Brain Res 1071: 218–225.
Franklin TR, Acton PD, Maldjian JA, Gray JD, Croft JR, Dackis CA et al (2002). Decreased gray matter concentration in the insular, orbitofrontal, cingulate and temporat cortices of cocaine patients. Biol Psychiatry 51: 134–142.
Fuchs RA, Branham RK, See RE (2006). Different neural substrates mediate cocaine seeking after abstinence vs extinction training: a critical role for the dorsolateral caudate-putamen. J Neurosci 26: 3584–3588. Critical demonstration that extinction training imposes a more enriched circuit into the relapse to drug-seeking.
Garavan H, Pankiewicz J, Bloom A, Cho JK, Sperry L, Ross TJ et al (2000). Cue-induced cocaine craving: neuroanatomical specificity for drug users and drug stimuli. Am J Psychiatry 157: 1789–1798.
Goldstein RA, Volkow ND (2002). Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 159: 1642–1652.
Gonzales RA, Weiss F (1998). Suppression of ethanol-reinforced behavior by naltrexone with attenuation of the ethanol-induced increase in dialysate dopamine levels in the nucleus accumbens. J Neuroscience 18: 10663–10671.
Graham DI, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW (2007). Dynamic BDNF activity in the nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neuroscience 10: 1029–1037.
Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y (2003). Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci 23: 742–747.
Guillin O, Diaz J, Carroll P, Griffon N, Schwartz JC, Sokoloff P (2001). BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 411: 86–89.
Hart CL, Haney M, Vosburg SK, Rubin E, Foltin RW (2007). Smoked Cocaine self-administration is decreased by Modafinil. Neuropsychopharmacology (advance online publication, 13 June 2007).
Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR (1999). Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci 19: 4110–4122.
Hurd YL, Herkenham M (1993). Molecular alterations in the neostriatum of human cocaine addicts. Synapse 13: 357–369.
Hyman SE, Malenka RC, Nestler EJ (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598.
Jay TM (2003). Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol 69: 375–390.
Jedynak JP, Uslaner JM, Esteban JA, Robinson TE (2007). Methamphetamine-induced structural plasticity in the dorsal striatum. Eur J Neurosci 25: 847–853.
Jentsch K, Taylor J (1999). Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacol 146: 373–390.
Jin SH, Blendy JA, Thomas SA (2005). Cyclic AMP response element-binding protein is required for normal maternal nurturing behavior. Neuroscience 133: 647–655.
Jones S, Bonci A (2005). Synaptic plasticity and drug addiction. Curr Opin Pharmacol 5: 20–25. This review nicely summarizes the role of the VTA in the transition between short and long-term neuroplasticity.
Kalivas PW, Hu XT (2006). Exciting inhibition in psychostimulant addiction. Trends Neurosci 29: 610–616.
Kalivas PW, Volkow N, Seamans J (2005). Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron 45: 647–650.
Kalivas PW, Volkow ND (2005). The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry 162: 1403–1413.
Kampman KM, Pettinati H, Lynch KG, Dakis C, Sparkman T, Weigley C et al (2004). A pilot trial of topiramate for the treatment of cocaine dependence. Drug Alcohol Depend 75: 233–240.
Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H (2003). Structure-stability-function relationships of dendritic spines. Trends Neurosci 26: 360–368.
Kelley AE (2004). Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron 44: 161–179. Very clear statement of the overlap between normal motivated learning and the development of addiction.
Kelz MB, Chen J, Carlezon Jr WA, Whisler K, Gilden L, Beckmann AM et al (1999). Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature 401: 272–276.
Kolb B, Pellis S, Robinson TE (2004). Plasticity and functions of the orbital frontal cortex. Brain Cogn 55: 104–115.
Kourrich S, Rothwell PE, Klug JR, Thomas MJ (2007). Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci 27: 7921–7928.
LaLumiere R, Kalivas P (2006). Reward and drugs of abuse. In: Kesner R, Martinez J (eds). Neurobiology of Memory and Learning, 2nd edn. Academic Press: New York. pp 459–483.
LaRowe S, Myrick H, Hedden S, Stroud Z, Mardikian P, Saladin M et al (2007). Cocaine desire is reduced by N-acetylcysteine. Am J Psychiatry 164: 1115–1117.
Laviolette SR, van der Kooy D (2004). The neurobiology of nicotine addiction: bridging the gap from molecules to behaviour. Nat Rev Neurosci 5: 55–65.
Le Foll B, Diaz J, Sokoloff P (2005). A single cocaine exposure increases BDNF and D3 receptor expression: implications for drug-conditioning. Neuroreport 16: 175–178.
Lippman J, Dunaevsky A (2005). Dendritic spine morphogenesis and plasticity. J Neurobiol 64: 47–57.
Lisman J (2003). Actin's actions in LTP-induced synapse growth. Neuron 38: 361–362.
Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y (2004b). A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci 24: 1604–1611.
Lu L, Grimm JW, Hope BT, Shaham Y (2004a). Incubation of cocaine craving after withdrawal: a review of preclinical data. Neuropharmacology 47 (Suppl 1): 214–226. Nice overview of the importance of neuroplasticity that progresses during abstinences and the behavioral consequences of progressive plasticity.
Lu L, Grimm JW, Shaham Y, Hope BT (2003). Molecular neuroadaptations in the accumbens and ventral tegmental area during the first 90 days of forced abstinence from cocaine self-administration in rats. J Neurochem 85: 1604–1613.
Mackler SA, Korutla L, Cha XY, Koebbe MJ, Fournier KM, Bowers MS et al (2000). NAC-1 is a brain POZ/BTB protein that can prevent cocaine-induced sensitization in the rat. J Neurosci 20: 6210–6217.
Malcolm R, Swayngim K, Donovan JL, DeVane CL, Elkashef A, Chiang N et al (2006). Modafinil and cocaine interactions. Am J Drug Alcohol Abuse 32: 577–587.
Mangiavacchi S, Wolf ME (2004). D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J Neurochem 88: 1261–1271.
Mansvelder HD, McGehee DS (2000). Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27: 349–357.
Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A (2006). Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci 9: 868–869.
Martinez D, Narendran R, Foltin RW, Slifstein M, Hwang D-R, Broft A et al (2007). Amphetamine-induced dopamine release is markedly blunted in cocaine dependence and predictive of the choice to self administer cocaine. Am J Psychiatry 164: 622–629.
Matus A (2005). Growth of dendritic spines: a continuing story. Curr Opin Neurobiol 15: 67–72.
McBean GJ (2002). Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci 23: 299–302.
McClung CA, Nestler EJ (2003). Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci 6: 1208–1215.
McFarland K, Davidge SB, Lapish CC, Kalivas PW (2004). Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci 24: 1551–1560.
McFarland K, Kalivas PW (2001). The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 21: 8655–8663.
McFarland K, Lapish CC, Kalivas PW (2003). Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23: 3531–3537.
McGinty JF (2007). Co-localization of GABA with other neuroactive substances in the basal ganglia. Prog Brain Res 160: 273–284.
Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M et al (2002). Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35: 121–133.
Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK (2005). Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci 25: 6389–6393.
Moratalla R, Elibol B, Vallejo M, Graybiel AM (1996). Network-level changes in expression of inducible fos-jun proteins in the striatum during chronic cocaine treatment and withdrawal. Neuron 17: 147–156.
Myrick H, Anton R (2004). Recent advances in the pharmacotherapy of alcoholism. Curr Psychiatry Rep 6: 332–338.
Nader K, van der Kooy D (1997). Deprivation state switches the neurobiological substrates mediating opiate reward in the ventral tegmental area. J Neurosci 17: 383–390.
Nader MA, Czoty PW (2005). PET imaging of dopamine D2 receptors in monkey models of cocaine abuse: genetic predisposition vs environmental modulation. Am J Psychiatry 162: 1473–1482.
Neisewander JL, Fuchs RA, Tran-Nguyen LT, Weber SM, Coffey GP, Joyce JN (2004). Increases in dopamine D3 receptor binding in rats receiving a cocaine challenge at various time points after cocaine self-administration: implications for cocaine-seeking behavior. Neuropsychopharmacology 29: 1479–1487.
Nestler E (2001). Molecular basis of long-term plasticity underlying addiction. Nature Rev 2: 119–128.
Nestler EJ (2005). Is there a common molecular pathway for addiction? Nat Neurosci 8: 1445–1449. Excellent overview of molecular plasticity produced by addictive drugs, with a focus on identifying plasticity that generalizes between drugs.
Nestler EJ, Barrot M, Self DW (2001). DeltaFosB: a sustained molecular switch for addiction. Proc Natl Acad Sci USA 98: 11042–11046.
Nicola SM, Surmeier J, Malenka RC (2000). Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci 23: 185–215.
Nogueira L, Kalivas PW, Lavin A (2006). Long-term neuroadaptations produced by withdrawal from repeated cocaine treatment: role of dopaminergic receptors in modulating cortical excitability. J Neurosci 26: 12308–12313.
Norrholm SD, Bibb JA, Nestler EJ, Ouimet CC, Taylor JR, Greengard P (2003). Cocaine-induced proliferation of dendritic spines in the nucleus accumbens is dependent on the activity of cyclin-dependent kinase-5. Neuroscience 116: 19–22.
O'Brien CP (1975). Experimental analysis of conditioning factors in human narcotic addiction. Pharmacol Rev 27: 533–543.
O'Brien CP (2003). Research advances in the understanding and treatment of addiction. Am J Addict 12 (Suppl 2): S36–S47.
O'Brien CP (2005). Anticraving medications for relapse prevention: a possible new class of psychoactive medications. Am J Psychiatry 162: 1423–1431.
O'Brien CP, Brady JP, Wells B (1977). Condictioned narcotic withdrawal in jumans. Science 195: 1000–1002.
O'Brien CP, Volkow N, Li TK (2006). What's in a word? Addiction vs dependence in DSM-V. Am J Psychiatry 163: 764–765.
Pandey SC, Roy A, Zhang H, Xu T (2004). Partial deletion of the cAMP response element-binding protein gene promotes alcohol-drinking behaviors. J Neurosci 24: 5022–5030.
Peters J, Kalivas PW (2006). The group II metabotropic glutamate receptor agonist, LY379268, inhibits both cocaine- and food-seeking behavior in rats. Psychopharmacology (Berl) 186: 143–149.
Pierce RC, Kalivas PW (1997). A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain ResRev 25: 192–216.
Pierce RC, Kumaresan V (2006). The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev 30: 215–238.
Pu L, Liu QS, Poo MM (2006). BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci 9: 605–607.
Rao A, Craig AM (2000). Signaling between the actin cytoskeleton and the postsynaptic density of dendritic spines. Hippocampus 10: 527–541.
Richards DA, Mateos JM, Hugel S, de Paola V, Caroni P, Gahwiler BH et al (2005). Glutamate induces the rapid formation of spine head protrusions in hippocampal slice cultures. Proc Natl Acad Sci USA 102: 6166–6171.
Rilling J, Gutman D, Zeh T, Pagnoni G, Berns G, Kilts C (2002). A neural basis for social cooperation. Neuron 35: 395–405.
Robinson TE, Kolb B (1999). Morphine alters the structure of neurons in the nucleus accumbens and neocortex of rats. Synapse 33: 160–162.
Robinson TE, Kolb B (2004). Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 47 (Suppl 1): 33–46. Outstanding overview of the changes in spine density associated with chronic use of addictive drugs.
Schultz W (1998). Predictive reward signal of dopamine neurons. Am J Physiol 80: 1–27.
Schultz W (2004). Neural coding of basic reward terms of animal learning theory, game theory, microeconomics and behavioural ecology. Curr Opin Neurobiol 14: 139–147.
See RE (2002). Neural substrates of conditioned-cued relapse to drug-seeking behavior. Pharmacol Biochem Behav 71: 517–529.
Seiden LS, Sabol KE, Ricuarte GA (1993). Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol 33: 639–677.
Sierra-Mercado Jr D, Corcoran KA, Lebron-Milad K, Quirk GJ (2006). Inactivation of the ventromedial prefrontal cortex reduces expression of conditioned fear and impairs subsequent recall of extinction. Eur J Neurosci 24: 1751–1758.
Soosairajah J, Maiti S, Wiggan O, Sarmiere P, Moussi N, Sarcevic B et al (2005). Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J 24: 473–486.
Steward O, Worley PF (2001). A cellular mechanism for targeting newly synthesized mRNAs to synaptic sites on dendrites. Proc Natl Acad Sci USA 98: 7062–7068.
Sun W, Rebec GV (2005). The role of prefrontal cortex D1-like and D2-like receptors in cocaine-seeking behavior in rats. Psychopharmacology (Berl) 177: 315–323.
Sun X, Zhao Y, Wolf ME (2005). Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci 25: 7342–7351.
Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D et al (2003). Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature 421: 70–75. Breakthrough study showing that extinction is an active learning process producing changes in prefrontal to accumbens glutamatergic transmission.
Szumlinski KK, Abernathy KE, Oleson EB, Klugmann M, Lominac KD, He DY et al (2006). Homer isoforms differentially regulate cocaine-induced neuroplasticity. Neuropsychopharmacology 31: 768–777.
Tang XC, McFarland K, Cagle S, Kalivas PW (2005). Cocaine-induced reinstatement requires endogenous stimulation of mu-opioid receptors in the ventral pallidum. J Neurosci 25: 4512–4520.
Toda S, Shen HW, Peters J, Cagle S, Kalivas PW (2006). Cocaine increases actin cycling: effects in the reinstatement model of drug seeking. J Neurosci 26: 1579–1587. Paper describing the role of increased actin cycling in regulating excitatory transmission in addiction.
Todtenkopf MS, Parsegian A, Naydenov A, Neve RL, Konradi C, Carlezon Jr WA (2006). Brain reward regulated by AMPA receptor subunits in nucleus accumbens shell. J Neurosci 26: 11665–11669.
Trantham H, Szumlinski K, McFarland K, Kalivas P, Lavin A (2002). Repeated cocaine administration alters the electrophysiological properties of prefrontal cortical neurons. Neuroscience 113: 749.
Vocci F, Ling W (2005). Medications development: successes and challenges. Pharmacol Ther 108: 94–108. Outstanding overview of the current status of many potential treatments for addiction.
Volkow ND, Fowler JS, Wang GJ, Swanson JM (2004). Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry 9: 557–569. This article describes the remarkable decrease in dopamine transmission in the striatum that accompanies addiction to a variety of drugs of abuse.
Volkow ND, Wang GJ, Fowler JS, Thanos PP, Logan J, Gatley SJ et al (2002). Brain DA D2 receptors predict reinforcing effects of stimulants in humans: replication study. Synapse 46: 79–82.
Volkow ND, Wang GJ, Ma Y, Fowler JS, Wong C, Ding YS et al (2005). Activation of orbital and medial prefrontal cortex by methylphenidate in cocaine-addicted subjects but not in controls: relevance to addiction. J Neurosci 25: 3932–3939.
Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Childress AR et al (2006). Cocaine cues and dopamine in the dorsal striatum: mechanism of craving in cocaine addiction. J Neuroscience 26: 6583–6588.
Volpicelli JR, Watson NT, King AC, Sherman CE, O'Brien CP (1995). Effect of naltrexone on alcohol ‘high’ in alcoholics. Am J Psychiatry 152: 613–615.
Walters CL, Cleck JN, Kuo YC, Blendy JA (2005). Mu-opioid receptor and CREB activation are required for nicotine reward. Neuron 46: 933–943.
Wilson SJ, Sayette MA, Fiez JA (2004). Prefrontal responses to drug cues: a neurocognitive analysis. Nat Neurosci 7: 211–214.
Wise RA (2004). Dopamine, learning and motivation. Nat Neuro Rev 5: 483–494.
Wolf ME, Sun X, Mangiavacchi S, Chao SZ (2004). Psychomotor stimulants and neuronal plasticity. Neuropharmacology 47 (Suppl 1): 61–79.
Xi ZX, Newman AH, Gilbert JG, Pak AC, Peng XQ, Ashby Jr CR et al (2006). The novel dopamine D3 receptor antagonist NGB 2904 inhibits cocaine's rewarding effects and cocaine-induced reinstatement of drug-seeking behavior in rats. Neuropsychopharmacology 31: 1393–1405.
Xi ZX, Ramamoorthy S, Baker DA, Shen H, Samuvel DJ, Kalivas PW (2002). Modulation of group II metabotropic glutamate receptor signaling by chronic cocaine. J Pharmacol Exp Ther 303: 608–615.
Yao L, McFarland K, Fan P, Jiang Z, Inoue Y, Diamond I (2005). Activator of G protein signaling 3 regulates opiate activation of protein kinase A signaling and relapse of heroin-seeking behavior. Proc Natl Acad Sci USA 102: 8746–8751.
Zachariou V, Bolanos CA, Selley DE, Theobald D, Cassidy MP, Kelz MB et al (2006). An essential role for DeltaFosB in the nucleus accumbens in morphine action. Nat Neuroscience 9: 205–211.
Zhang XF, Hu XT, White FJ (1998). Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci 18: 488–498.
Acknowledgements
This work was supported in part by USPHS Center Grants P50 DA015369 and P60 DA01586.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE
Dr O'Brien has served as a consultant in the past three years to Alkermes, Cephalon, Forest and McNeil Laboratories. Dr Kalivas has nothing to disclose.
Rights and permissions
About this article
Cite this article
Kalivas, P., O'Brien, C. Drug Addiction as a Pathology of Staged Neuroplasticity. Neuropsychopharmacol 33, 166–180 (2008). https://doi.org/10.1038/sj.npp.1301564
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301564
Keywords
This article is cited by
-
Net gain and loss: influence of natural rewards and drugs of abuse on perineuronal nets
Neuropsychopharmacology (2023)
-
Synaptic plasticity and mental health: methods, challenges and opportunities
Neuropsychopharmacology (2023)
-
Plasma endocannabinoids in cocaine dependence and their relation to cerebral metabotropic glutamate receptor 5 density
Translational Psychiatry (2023)
-
Cocaine induces input and cell-type-specific synaptic plasticity in ventral pallidum-projecting nucleus accumbens medium spiny neurons
Neuropsychopharmacology (2022)
-
Inhibition of a cortico-thalamic circuit attenuates cue-induced reinstatement of drug-seeking behavior in “relapse prone” male rats
Psychopharmacology (2022)
