
Abstract
Decreased synaptic serotonin during depressive episodes is a central element of the monoamine hypothesis of depression. The serotonin transporter (5-HTT, SERT) is a key molecule for the control of synaptic serotonin levels. Here we aimed to detect state-related alterations in the efficiency of 5-HTT-mediated inward and outward transport in platelets of drug-free depressed patients suffering from seasonal affective disorder (SAD). 5-HTT turnover rate, a measure for the number of inward transport events per minute, and tyramine-induced, 5-HTT-mediated outward transport were assessed at baseline, after 4 weeks of bright light therapy, and in summer using a case–control design in a consecutive sample of 73 drug-free depressed patients with SAD and 70 nonseasonal healthy controls. Patients were drug-naive or medication-free for at least 6 months prior to study inclusion, females patients were studied in the follicular phase of the menstrual cycle. All participants were genotyped for a 5-HTT-promoter polymorphism (5-HTTLPR) to assess the influence of this polymorphism on 5-HTT parameters. Efficiency of 5-HTT-mediated inward (p=0.014) and outward (p=0.003) transport was enhanced in depressed patients. Both measures normalized toward control levels after therapy and in natural summer remission. Changes in outward transport showed a clear correlation with treatment response (ρ=0.421, p=0.001). Changes in inward transport were mediated by changes in 5-HTT transport efficiency rather than affinity or density. 5-HTTLPR was not associated with any of the 5-HTT parameters. In sum, we conclude that the 5-HTT is in a hyperfunctional state during depression in SAD and normalizes after light therapy and in natural summer remission.
Similar content being viewed by others
INTRODUCTION
A basic tenet of the monoamine hypothesis of depression is that synaptic serotonin is low during major depressive episodes. Lowering of the serotonin precursor tryptophan by dietary tryptophan depletion causes a 30–60% decrease in brain serotonin content in animals and a transient reappearance of depressive symptoms in remitted patients with seasonal (Lam et al, 1996; Neumeister et al, 1997, 1998) and nonseasonal depression (Delgado et al, 1991; Smith et al, 1997). A decrease in synaptic serotonin in depression is further suggested by challenge studies with serotonergic agents (Levitan et al, 1998; Schwartz et al, 1997). Still, the literature proposes few explanations on how the supposed reduction in synaptic serotonin may arise.
The main mechanism controlling synaptic serotonin levels is Na+/Cl-dependent reuptake into the presynaptic neuron via the 5-HT-transporter (5-HTT, serotonin transporter (SERT); Rudnick and Clark, 1993). Transport can be reversed by manipulating ion gradients (Pifl and Singer, 1999; Sitte et al, 2001) or by amphetamine-like substances such as tyramine (TYR; Hilber et al, 2005; Schuemann, 1960). The 5-HTT is expressed in the central nervous system (CNS) and peripheral tissues, including blood platelets. It is encoded by a single gene (Lesch et al, 1993; Ramamoorthy et al, 1993) regulated by a polymorphic promoter region (5-HTT-promoter polymorphism, 5-HTTLPR (Heils et al, 1996; Lesch et al, 1996). Brain imaging techniques (positron emission tomography or single photon computed tomography, SPECT) allow for measurement of 5-HTT availability in the living human brain. While some studies suggest reductions in CNS 5-HTT availability in seasonal (Willeit et al, 2000) and nonseasonal depression (Malison et al, 1998; Parsey et al, 2006; Staley et al, 2006), recent studies failed to find altered 5-HTT binding (Meyer et al, 2004). An important limitation of current brain imaging techniques is that they are confined to quantification of 5-HTT binding sites but do not allow for functional assessments of transport processes.
Investigating maximal binding capacity (Bmax) of radiolabeled ligands to platelet 5-HTTs is a classical research paradigm in biological psychiatry. Most studies did not screen for seasonality, and results vary, partly due to methodological differences. Studies using [3H]imipramine suggest reduced 5-HTT expression during depression (Owens and Nemeroff, 1994), while binding of radiolabeled selective serotonin reuptake inhibitors seems to be unaltered (D'Hondt et al, 1994; Rosel et al, 1999) or increased (Neuger et al, 1999). A minority of studies (eg Franke et al, 2000, 2003; Neuger et al, 1999; Stain-Malmgren et al, 1998) investigated maximal transport velocity (Vmax) in nonseasonal depression, with some of them (Neuger et al, 1999) reporting decreased Vmax in female patients. Few investigated Vmax in direct comparison to Bmax (Nobile et al, 1999). A study in patients with seasonal affective disorder (SAD) showed reduced Bmax of [3H]paroxetine in patients with SAD, but no significant differences in Vmax (Stain-Malmgren et al, 1998). To our knowledge, as of yet there is no study investigating pharmacologically induced, 5-HTT-mediated outward transport in depression.
This study investigated 5-HTT function in SAD, winter-type (Rosenthal et al, 1984), a subform of recurrent major depression (DSM-IV, 1994) where depressive episodes during fall/winter alternate with remission or hypomania during spring/summer. Bright light therapy (BLT) is a biologically active first-line treatment for SAD (Avery, 1998; Eastman et al, 1998; Lam et al, 2006; Lewy et al, 1998; Terman et al, 1998; Wirz-Justice, 1998). Since BLT, in contrast to pharmacotherapy, does not directly interfere with 5-HTT function, it offers the opportunity to investigate antidepressant response at the level of 5-HTT without interfering with measurements of 5-HTT parameters.
The two main functional parameters assessed in the present study are 5-HTT-mediated inward and outward transport. Inward transport was assessed by measuring 5-HTT turnover rates, which represent the number of transport events occurring at a 5-HTT molecule per min. Outward transport was assessed by measuring TYR-induced outward transport (ETYR) of the 5-HTT-substrate [3H]1-methyl-4-phenylpyridinium (MPP+; Cesura et al, 1987; Javitch et al, 1985). With the exception of pharmacological events associated with stimulant use and abuse, we are not aware of any role for 5-HTT-mediated outward transport in the human brain. However, inward and outward transport provide information on the efficiency of 5-HTT-mediated transmembrane transport. These parameters were determined in drug-free patients with SAD and in healthy control subjects at baseline, after BLT, and in summer.
In view of the evidence for reduced serotonin levels during depression, and the antidepressant effect of 5-HTT blockade, we hypothesized that compared to healthy controls, patients with SAD would display higher 5-HTT turnover rates. Since outward transport is an in vitro probe for 5-HTT transport efficiency, we hypothesized that ETYR will be enhanced in SAD patients. Our secondary hypothesis was that remission of depressive symptoms would be associated with a normalization of transport parameters toward control levels. In replication of earlier work (Greenberg et al, 1999), we further hypothesized that carriers of the 5-HTTLPR long allele would display increased Vmax values and significant seasonal variations in Vmax.
METHODS
Study Sample
The study and recruitment procedures were approved by the Ethics Committee of the Medical University Vienna (MUV). All participants gave written informed consent after full explanation of study procedures.
Patients were recruited at the Outpatient Clinic for SAD, Department of Biological Psychiatry, MUV, after self-referral or referral through their treating psychiatrists or general practitioners. Demographic and clinical information about longitudinal course of illness, previous treatment attempts, and other psychiatric diagnoses was obtained using a semi-structured interview based on DSM-IV (First et al, 1996), and by review of medical records and direct contact with previous psychiatrists. Patients were diagnosed by consensus of the authors using DSM-IV criteria for SAD. All patients were either drug-naive or free of psychotropic medication for at least 6 months prior to study inclusion. All study subjects underwent medical screening including physical examination, medical history, and routine hematology. Patients with a history of substance abuse 6 months prior to inclusion, or DSM-IV Axis I disorders other than SAD were not included into the study.
Healthy volunteers were recruited by newspaper advertisements and word of mouth. To avoid selection bias, information about precise study aims was withheld during pre-inclusion phase. The absence of any past or present DSM-IV Axis I disorder was ascertained using the Structured Clinical Interview for DSM-IV nonpatient version (First et al, 1996) administered by an experienced rater (NT) and the Structured Interview Guide for the Hamilton Depression Rating Scale (HDRS) (Hamilton, 1967), Seasonal Affective Disorder Version (SIGH-SAD) (Williams et al, 1988) consisting of the HDRS and the supplement for atypical depressive symptoms. Control subjects had a negative family history for axis I disorders as ascertained by the family history screen (Weissman et al, 2000).
Patients and control subjects were given a German version of the Seasonal Pattern Assessment Questionnaire (Kasper, 1991) to assess the Global Seasonality Score (GSS). Control subjects with a GSS>6 were not included. Within each study year (fall to summer), patients and control subjects were matched as a group for gender and age. For every patient entering the study, a control subject was included within 2 weeks. To minimize variability introduced by the influence of the menstrual cycle on 5-HTT function (Maswood et al, 1999), all premenopausal female patients had their visits in the follicular phase of their menstrual cycle. The entire study sample was of Caucasian origin.
Psychopathological Measurements
SIGH-SAD ratings were performed at baseline (first blood sampling), after 2 weeks, after 4 weeks BLT (second blood sampling), and within 4 weeks of 180 days after baseline visit during natural summer remission (third blood sampling). Only patients attaining a baseline SIGH-SAD score of 20 or more were included in the study. All psychopathological ratings were carried out by one and the same experienced psychiatrist (NT).
Bright Light Therapy
All participants were provided with standard BLT devices delivering white light in an intensity of 10 000 lux at a distance of approximately 60 cm. They were instructed to undergo 45 min of BLT shortly after arising from bed. To ensure compliance, all participants were asked to fill out a daily protocol on time and duration of their BLT sessions. Full remission was defined as a maximum posttreatment SIGH-SAD score of 8.
Pharmacological Measures
Blood withdrawal and sample handling
Blood samples (90 ml) were collected by venipuncture (EDTA tubes, 1% wt/vol in saline). All samples were anonymized and prepared for functional 5-HTT testing within 1 h from withdrawal. To control for possible circadian variations in 5-HTT function (Rausch et al, 2005), all blood samples were drawn between 8 and 9 a.m. throughout the study. All laboratory staff was fully blinded to the subject's status.
Platelet-rich plasma (PRP) was separated from blood cells by centrifugation (223g, Beckman-JS-21, 22°C) and diluted with CO2-gassed Krebs–Henseleit buffer (KH; 6.92 g NaCl, 0.35 g KCl, 0.29 g MgSO4 × 7H2O, 0.16 g KH2PO4, 2.1 g NaHCO3, 2.1 g glucose per liter, pH=7.4). Subsequently, PRP was used for uptake and superfusion experiments, and membranes were prepared for binding experiments. Platelets were isolated from supernatant by centrifugation (1470g, Sorvall-GLC-3 centrifuge, 15 min, swing-out rotor, 4°C). The supernatant was recovered as platelet-free plasma (PFP) and re-centrifuged using conditions as above. Platelet pellets were resuspended in KH containing 10% PFP and used in uptake and outward transport assays at 37°C. All experiments were done in triplicate determination.
Inward transport
For uptake assays, resuspended platelet solution (50 μl) was incubated for 3 min with various (5-HT) (0.03, 0.1, 0.3, 1.3, 10 μM unlabeled and [3H]5-HT (Scholze et al, 2001); specific activity 21.5–25.8 Ci/mmol; constant (0.03 μM), 500 μl KH). Nonspecific uptake was determined at 10 μM serotonin in the presence of 1 μM paroxetine. Uptake was assessed by using a dilution technique with unlabeled 5-HT to reveal Vmax and Km values (by recalculating and fitting the background-corrected uptake data to Michaelis–Menten kinetics, with c.p.m. values at the highest [5-HT] being 3–9 times over background). Uptake reaction was stopped as described above.
Binding experiments, turnover rates, and blood 5-HT content
For binding experiments, PRP was centrifuged (1659g, 15 min, Beckman-JS-21, swing-out rotor, room temperature), plasma was removed subsequently. Platelet pellets were stored at −80°C for up to 2 months. Pellets were resuspended in 3 ml of KH (without glucose) and centrifuged (19 510g, 10 min, 4°C) and resuspended in 1 ml ice-cold HME buffer (25 mM HEPES NaOH (pH 7.5), 2 mM MgCl2, 1 mM EDTA) and centrifuged (19 510g, 10 min, 4°C). The pellet was resuspended in 0.5 ml HME (4°C) and subjected to a freeze-thaw cycle (liquid nitrogen) with subsequent sonication (40%, 20 pulses). Membranes were centrifuged (19 510g, 10 min, 4°C). Pellets were resuspended in KH (stored after snap freezing in liquid nitrogen, −80°C). Membranes were incubated in KH (500 μl, 22°C) containing [3H]β-CIT ((1R,2S,3S,5S)-3-(4-iodophenyl)-8-[3H]methyl-8-azabicyclo[3.2.1]octane-2-carboxylic acid; 0.01, 0.03, 0.1, 0.3, 1.3, 10 nM, specific activity 60– 80 Ci/mmol). Nonspecific binding was determined in the presence of 3 μM paroxetine. After 60 min, 1 ml ice-cold KH was added and filtration through Whatman GF/C-filters at 4°C (presoaked with 0.3% polyethyleneimine) performed. Protein concentration of platelet samples was measured with a Pierce BCA kit (Pierce, Rockford, IL); Vmax and Bmax values were normalized to the protein content of the platelet sample. To obtain turnover rates, Vmax of [3H]serotonin and Bmax of [3H]β-CIT were determined in separate, though parallel measurements. Blood 5-HT content in PRP and PFP was determined using an ELISA kit (RE 591 21; IBL Hamburg, Germany). In brief, a 50 μl sample was acylated, centrifuged, and incubated together with control plasma and standards into 96-well plates. A total of 50 μl 5-HT biotin and 50 μl anti-sera were mixed and incubated overnight (4°C). Enzyme conjugate and p-nitrophenyl-phosphate (PNPP) substrate solution were added and incubated for 1 h. The reaction was stopped with PNPP stop solution. Optical density was then measured using an Anthos Elisa plate reader at 405 nm.
Outward transport
Resuspended platelets were incubated with [3H]MPP+ for 1 h. In contrast to [3H]5-HT, [3H]MPP+ is neither metabolized nor diffusible and thus gives a high signal-to-noise ratio (Scholze et al, 2001). Subsequently, platelet solution was re-centrifuged using conditions as described above, washed with prewarmed KH to remove residual radioactivity, and resuspended in KH. Aliquots were used in outward transport assays. Efflux was induced by addition of TYR, and to ensure the specific, 5-HTT-mediated nature of the efflux, by addition of the Na+/H+ ionophore monensin (EMON) (Scholze et al, 2000), and a mixture of both (ETYR/MON; final concentrations 100 and 1 μM, respectively). Reactions were stopped by addition of 1 ml ice-cold KH and immediate centrifugation (4°C, Sorvall-GLC-3, 1470g).
Data calculation for experimentally determined values
Data were adjusted for blank values and fitted to hyperbolic saturation isotherms (uptake, binding) using Prism (GraphPad, San Diego, CA). Turnover rates were calculated by dividing Vmax by Bmax in each sample (1 per min).
Genotyping
Genomic DNA was extracted from leukocytes or whole blood according to standard procedures. Polymerase chain reaction amplification was performed by using the primers and procedures described by Cook et al (1997). PCR products were separated on 3% agarose gels and visualized by ethidium bromide staining.
Chemicals
Chemicals were obtained from following the companies: [3H]MPP+ and [3H]serotonin, PerkinElmer Life Sciences Products (Boston, MA); [3H]β-CIT, Amersham Biosciences Europe GMBH (Vienna, Austria); TYR, monensin and serotonin, Sigma-Aldrich Handels GmbH (Vienna, Austria); MPP+, RBI/Sigma (Natick, MA); Paroxetine-HCl was a generous gift from GlaxoSmithKline Pharma GmbH, Vienna, Austria. All other chemicals were from commercial sources.
Statistical Analysis
Nonparametric tests were used since some of the variables did not meet assumption of normal distribution at some time points. Group differences between matched patients and control pairs and within-subject changes after BLT were assessed using Wilcoxon rank-sum tests and the corresponding 95% confidence intervals (CIs) in the location parameter. Circannual changes in 5-HTT parameters (before BLT, after BLT, summer) were tested for significance using Friedman tests for related variables. Nonparametric correlations were calculated between 5-HTT parameters and psychopathological ratings. Mann–Whitney U-tests were used to compare patients who remitted on BLT to BLT nonresponders. Wilcoxon signed-ranks tests were used for a group analysis of changes in 5-HTT parameters in BLT full-, partial-, and nonresponders. To exclude a significant influence of sex and age (Neuger et al, 1999; Staley et al, 2006) onto the main findings, an analysis of variance (ANOVA) was calculated for the two main outcome parameters before BLT using sex and age as covariates. Adjustment for multiple testing by the factor 2 was applied for the two main outcome measures (ETYR and 5-HTT turnover rate), p-values below 0.025 were considered to be statistically significant.
RESULTS
Study Sample and Response to Treatment
Of 415 patients visiting at the outpatient clinic for SAD between fall/winter 1999 and spring 2002, 251 met DSM-IV criteria for SAD. Of these patients, 178 did not meet inclusion criteria because they were (1) taking psychotropic medication at inclusion or within 6 months prior to inclusion (136); (2) had relevant comorbid medical, neurological, or psychiatric conditions (31); (3) or were unwilling to consent to the study protocol (11). Data of 73 patients (49 females, 24 males, age 38.70±13.1 years, mean±SD) and 70 healthy controls (50 females, 20 males, age 39.23±13.4 years) entered final analysis. Six patients dropped out before study visit 2, leaving 67 for analysis at week 4. Another 16 dropped out before visit 3, leaving 51 patients who completed the full protocol. Reasons for premature study termination were initiation of antidepressant drug therapy (n=18), antihypertensive/antiaggregant therapy (n=1), and withdrawal of consent (n=3). All but two control subjects completed the full protocol.
After 4 weeks BLT, SIGH-SAD scores dropped from 25.71±5.0 to 11.33±6.9 (paired sample t-test: two-tailed t=14.8, d.f.=66, p<0.001) in patients with SAD and, in line with earlier findings (Partonen and Lonnqvist, 2000), from 0.71±1.2 to 0.44±1.0 in healthy controls (t=2.29, d.f.=69, p<0.025). In summer, SIGH-SAD scores dropped to 3.43±4.1 in patients with SAD (week 4 vs summer: t=7.23, d.f.=50, p<0.001) and to 0.18±0.4 in controls (t=2.43, d.f.=67, p<0.018).
Criteria for full remission were met by 27 patients after 4 weeks of BLT, another 19 patients showed reductions in SIGH-SAD scores of more than 50%. Twenty-one patients did not experience relevant changes in SIGH-SAD scores and were classified as BLT nonresponders.
Primary Outcome Measures
5-HTT turnover rates
Overall 5-HTT turnover rates were in good agreement with previously published data (Gu et al, 1994). At baseline, patients with SAD had significantly higher baseline 5-HTT turnover rates than healthy controls. No significant differences between patients and controls were seen after BLT or in summer. Turnover rates showed significant variation over the three time points in patients but not in controls (Table 1, Figure 1a). An ANOVA confirmed group differences between patients and controls (F(1)=8.228, p=0.005) but failed to show significant effects of sex (F(1)=2.565, p=0.112) or age (F(1)=0.124; p=0.725).

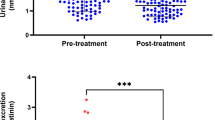
(a) Serotonin transporter (5-HTT) turnover rates in patients with SAD (n=61 before bright light therapy, BLT) and healthy control subjects before and after 4 weeks of BLT and in natural summer remission (SAD: empty squares, n=61 before BLT; healthy control subjects: filled diamonds, n=66 before BLT). * Group comparison between patients with SAD and healthy control subjects before BLT; Wilcoxon rank-sum test, p=0.014. (b) TYR-induced [3H]MPP+ outward transport (ETYR) in platelets of patients with SAD and healthy subjects before and after 4 weeks of BLT and in summer (SAD: n=66 before BLT; healthy control subjects: n=66 before BLT; legend same as in graph (a)). ** Group comparison between patients with SAD and healthy control subjects before BLT; Wilcoxon rank-sum test, p=0.003. (c) Maximal [3H]serotonin uptake capacity (Vmax) in platelets of patients with SAD (n=64 before BLT) and healthy controls (n=68 before BLT; legend same as in graph (a)). ** Group comparison before BLT; Wilcoxon rank-sum test, p=0.008. (d) Km of [3H]serotonin uptake in platelets of patients with SAD (n=64 before BLT) and healthy controls (n=68 before BLT; legend same as in graph (a)). (e) Maximal binding capacity (Bmax) of the 5-HTT ligand [3H]β-CIT in patients with SAD (n=63 before BLT) and healthy controls (n=67 before BLT; legend same as in graph (a)). (f) Kd of [3H]β-CIT binding to platelets in patients with SAD (n=63 before BLT) and healthy controls (n=67 before BLT; legend same as in graph (a)).
E TYR
At baseline, patients with SAD displayed significantly higher ETYR than control subjects. No significant group differences were seen after BLT and in summer. There were no significant fluctuations in ETYR over the three time points in either patients or controls (Table 1, Figure 1b). An ANOVA performed to exclude significant effects of sex and age onto the findings confirmed group differences in ETYR between patients and controls (F(1)=9.552, p=0.002) but failed to show significant effects of sex (F(1)=0.388, p=0.535) and age (F(1)=0.020; p=0.889).
Secondary Outcome Measures
[3H]5-HT uptake velocity and [3H]β-CIT-binding capacity
For a more detailed characterization of group differences and seasonal variation in 5-HTT turnover rates, separate post hoc analyses of [3H]serotonin uptake and [3H]β-CIT-binding experiments were performed.
V max
Compared to healthy controls, patients had significantly elevated values for maximal [3H]5-HT uptake velocity at baseline (mean Vmax (pmol/μg/min) 5.76 (n=64) vs 4.86 (n=68), 95% CIdiff 1.78–0.21, Wilcoxon rank-sum test: p=0.008). There were no significant differences in Vmax after BLT (3.50 (n=63) vs 2.98 (n=64), 95% CIdiff 1.13–0.32, p=0.28) or in summer (2.04 (n=43) vs 2.58 (n=64), 95% CIdiff 0.31–0.84, p=0.41). Vmax showed significant variation over the three time points in patients (Friedman's χ2(2)=21.714, p<0.001) and in healthy controls (Friedman's χ2(2)=17.803, p<0.001; Figure 1c).
No significant group differences were found in Km (Figure 1d), a correlate to the 5HT concentration at which the transport rate V=Vmax/2 and thereby a measure for the apparent substrate affinity (mean Km (μM) baseline: 0.303 (n=64) vs 0.232 (n=68), 95% CIdiff 0.05–0.003, p=0.08; after BLT: 0.335 (n=63) vs 0.306 (n=64), 95% CIdiff 0.07–0.01, p=0.11; summer: 0.305 (n=43) vs 0.232 (n=64), 95% CIdiff 0.05–0.002, p=0.32).
B max
There were no significant differences between patients and controls in maximal [3H]β-CIT-binding capacity at any point in time (mean Bmax (pmol/mg) baseline: 25.6 (n=63) vs 26.1 (n=67), 95% CIdiff 4.1–5.1; Wilcoxon rank-sum test: p=0.91; after BLT: 26.6 (n=63) vs 25.9 (n=65), 95% CIdiff 3.8–4.2, p=0.84; summer: 25.2 (n=44) vs 24.9 (n=64), 95% CIdiff 4.2–2.8, p=0.69; Figure 1e). Likewise, no significant group differences were found in 5-HTT affinity for [3H]β-CIT (mean Kd (nM) baseline: 0.245 (n=63) vs 0.246 (n=67), 95% CIdiff 0.05–0.03, p=0.77; after BLT: 0.251 (n=63) vs 0.237 (n=65), 95% CIdiff 0.06–0.03, p=0.52; summer: 0.221 (n=44) vs 0.260 (n=64), 95% CIdiff 0.02–0.04, p=0.38; Figure 1f). Bmax did not vary significantly over the three time points in patients (Friedman's χ2(2)=4.512, p=0.105) or healthy controls (Friedman's χ2(2)=2.381, p=0.304).
EMON/ETYR/MON
In both groups, magnitude of monensin-induced MPP+ outward transport was similar to that induced by TYR. Combined TYR/monensin-induced outward transport was approximately 50% higher than either ETYR or EMON. This is in good agreement with a specific, carrier-mediated, saturable outward transport through the 5-HTT. Although effect sizes were somewhat smaller for EMON and ETYR/MON, the pattern of alteration was similar in all three groups of outward transport experiments. At baseline, EMON was significantly elevated in patients compared to controls (mean EMON (%): 55.95 (n=66) vs 49.06 (n=66), 95% CIdiff 13.9–3.4, Wilcoxon rank-sum test: p=0.04). ETYR/MON was trend-wise elevated (mean ETYR/MON (%): 75.67 (n=68) vs 70.54 (n=65), 95% CIdiff 9.1–0.1, p=0.052). There were no significant differences between patients and controls after BLT (mean EMON: 52.96 (n=58) vs 55.66 (n=66), 95% CIdiff 3.1–9.0, p=0.35; mean ETYR/MON: 76.58 (n=57) vs 74.28 (n=67), 95% CIdiff 5.5–1.3, p=0.19) or in summer (mean EMON: 54.54 (n=46) vs 53.33 (n=65), 95% CIdiff 7.4–6.5, p=0.98; mean ETYR/MON: 80.93 (n=46) vs 77.53 (n=63), 95% CIdiff 5.3–2.6, p=0.55).
Blood 5-HT levels
Blood 5-HT levels were a good reflection of 5-HTT efficiency measures, with highest platelet 5-HT levels in depressed patients with SAD, and low levels in summer. 5-HT content in PFP showed the opposite pattern. However, differences between patients and controls did not reach significance for PRP 5-HT content (mean PRP 5-HT content (μg/μgProt) baseline: 6.7 (n=61) vs 5.4 (n=68), 95% CIdiff −0.9–3.4, Wilcoxon rank-sum test: p=0.08; after BLT: 5.9 (n=61) vs 5.0 (n=66), 95% CIdiff −0.9–2.8, p=0.44; summer: 4.0 (n=40) vs 4.8 (n=57), 95% CIdiff −2.1–0.4, p=0.51) and PFP 5-HT content (mean PFP 5-HT content (ng/ml) baseline: 9.5 (n=65) vs 11.4 (n=66), 95% CIdiff −4.6–0.9, p=0.24; after BLT: 9.3 (n=61) vs 11.8 (n=64), 95% CIdiff −5.5–0.5, p=0.12; summer: 11.4 (n=47) vs 12.9 (n=62), 95% CIdiff −5.6–2.5, p=0.31). In good agreement with seasonal changes in 5-HTT transport efficiency, PRP 5-HT content showed significant seasonal fluctuations both in patients (Friedman's χ2(2)=13.1, p=0.001) and controls (Friedman's χ2(2)=11.2, p=0.004).
Response to Treatment
To assess a possible relationship between 5-HTT function and response to BLT, rank correlations were calculated between posttreatment SIGH-SAD scores and pre- and posttreatment ETYR and 5-HTT turnover rates in patients with SAD. While no significant correlations were found between pretreatment 5-HTT parameters and posttreatment SIGH-SAD scores, both, posttreatment ETYR (Spearman's ρ: 0.392, p=0.002, n=58) and 5-HTT turnover rates (Spearman's ρ: 0.296, p=0.021, n=61) correlated positively with depression ratings after BLT, with lower posttreatment ETYR and lower 5-HTT turnover rates being associated with better response to BLT. There was also a significant correlation between relative reductions in SIGH-SAD scores calculated as ((pretreatment SIGH-SAD−posttreatment SIGH-SAD)/pretreatment SIGH-SAD) × 100) and relative changes in ETYR (calculated as above; Spearman's ρ: 0.421, p=0.001, n=58), indicating greater ETYR reductions in patients with better response to BLT. This was confirmed in a group analysis showing a drop in ETYR in BLT responders (Wilcoxon signed-ranks test: z=−2.0, p=0.046, n=24), but not in partial (z=−0.57, p=0.57, n=15) or nonresponders (z=−0.12, p=0.91, n=17; healthy controls showed an increase in ETYR after BLT; z=−2.584, p=0.01, n=64).
Posttreatment 5-HTT turnover rates in full responders were lower than in BLT nonresponders (21.39±21.4, n=26 vs 29.76±18.9, n=19; Mann–Whitney U-test, z=−2.298, p=0.022; Figure 2a). Similar results were obtained for ETYR (43.82±11.9, n=25 vs 52.72±11.8, n=17; Mann–Whitney U-test, z=−2.178, p=0.029; Figure 2b). No significant differences between remitters and nonresponders were found at baseline and in summer (Figure 2a and b; data not shown).

(a) Serotonin transporter (5-HTT) turnover rates in a subgroup of patients achieving full remission of depressive symptoms after 4 weeks of BLT (empty triangles; n=26 after BLT) and in BLT nonresponders (filled triangles; n=19 after BLT). * Group comparison after BLT; Mann–Whitney U-test, p=0.022. (b) TYR-induced [3H]MPP+ outward transport (ETYR) in a subgroup of patients achieving full remission of depressive symptoms after BLT (n=25 after BLT) and in BLT nonresponders (n=17 after BLT; legend same as in graph (a)). * Group comparison after BLT; Mann–Whitney U-test, p=0.029.
5-HTTLPR and 5-HTT Function
Genotype distribution did not deviate significantly from Hardy–Weinberg equilibrium in patients (χ2(1)=0.24, p>0.05) or controls (χ2(1)=0.28, p>0.05). In accordance with previous results (Johansson et al, 2003; Willeit et al, 2003), 5-HTTLPR genotype distribution did not differ significantly between patients with SAD and healthy controls (patients: ll: 22; ls: 38; ss: 13; controls: ll: 26; ls: 35; ss: 9; χ2(2)=1.121; p=0.571). An earlier study reported significant seasonal variations in serotonin uptake velocity in 5-HTTLPR ll homozygous subjects only (Greenberg et al, 1999). To replicate and extend this finding to other 5-HTT parameters, we performed analyses of variance separately in patients and controls using ETYR, 5-HTT turnover rates, Bmax, Vmax, and 5-HT levels in PRP and PFP as dependent variables and season (baseline vs summer) and the three genotypic groups as fixed factors. While the above-described influence of season on 5-HTT turnover rates and maximal 5-HT uptake velocity was confirmed in this analysis, the factor genotype (see also Lim et al, 2006) and the interaction terms genotype × season did not reach significance in any of the analyses (data not shown).
DISCUSSION
In this study, we provide, for the first time to the best of our knowledge, a comprehensive measurement of the forward and reverse 5-HTT transport modes in SAD. The present data show that 5-HTT turnover rates were elevated during depression, reduced toward control values after BLT, and slightly below control values in natural summer remission (Figure 1a). Likewise, TYR-induced outward transport of MPP+ via the 5-HTT is elevated in platelets of depressed patients with SAD (Figure 1b). Patients and control subjects did not differ anymore in ETYR after 4 weeks of BLT and in natural summer remission (Table 2).
Separate analysis of Vmax and Bmax, the two parameters used to determine 5-HTT turnover rates, showed that changes in serotonin uptake were mainly due to changes in Vmax (Figure 1c). Bmax, a measure for the amount of 5-HTT expressed in platelets, did not differ significantly between patients and control subjects at baseline, and it remained remarkably stable over all three time points in both groups (Figure 1e). Our methodology was not suited to discriminate 5-HTTs expressed on the platelet surface from intracellularly retained transporters. However, as shown by recent evidence, the preparation method used to produce platelet membranes reveals cell surface 5-HTTs rather than intracellularly retained 5-HTTs (Carneiro and Blakely, 2006). There were significant correlations between 5-HTT turnover numbers and ETYR at baseline (ρ=0.271, p=0.003) but not at later time points (after BLT: ρ=0.106, p=0.254; summer: ρ=−0.034, p=0.737). This suggests that efficiency of TYR uptake via the 5-HTT contributes to efflux measures, although not as the sole determinant.
Pretreatment 5-HTT turnover rates were highest in BLT nonresponders, somewhat lower in BLT responders, and lowest in healthy control subjects (Figures 1a and 2a). Patients who fully responded to treatment showed a drop in ETYR after 4 weeks of BLT, while nonresponders showed virtually no change in ETYR after BLT (Figure 2b). There were significant positive correlations between posttreatment SIGH-SAD scores and ETYR and 5-HTT turnover rates, and significant positive correlations between changes in ETYR and reductions in depression ratings after BLT. Before BLT, 5-HTT turnover numbers and ETYR correlated significantly in patients achieving full remission (ρ=0.624, p=0.002), at a trend level in partial responders (ρ=0.482, p=0.069), while there was no correlation in nonresponders (ρ=0.083, p=0.729). The 5-HTTLPR polymorphism was not associated with any of the functional 5-HTT parameters, and in contrast to some (Greenberg et al, 1999; Patkar et al, 2003), but not other findings (Javors et al, 2005), our data failed to show an influence of 5-HTTLPR on seasonal variations in Vmax or other 5-HTT parameters.
The factors influencing alterations in ETYR in patients with SAD are currently unknown. With a posttreatment drop in BLT responders, an increase after BLT in healthy controls, and no change in BLT nonresponders (Figures 1b and 2b), changes in ETYR may be the reflection of a compensatory process that BLT responders apply successfully, healthy controls do not need, and BLT nonresponders are not able to apply in response to physiological changes induced by exposure to BLT. However, elevated ETYR in our study may well reflect the measured enhancement of inward transport capacity, since current models explaining outward transport also assume a stoichiometrical coupling of the two transport modes (Fischer and Cho, 1979; Seidel et al, 2005). With all likelihood, humoral factors in peripheral blood acting either directly on platelets or at their precursor cells, the megakcariocytes, may account for the changes in platelet 5-HTT function. Factors previously suggested to influence 5-HTT function are—among others—steroids, especially β-estradiol (Chang and Chang, 1999). Endogenous TYR may interact with the 5-HTT under physiological conditions as it has been shown to mediate physiological changes in serotonin-related functions (Nisimura et al, 2005), and the literature offers numerous reports on alterations in TYR metabolism (Hale et al, 1989; Harrison et al, 1984; Sandler et al, 1975) and its relation to treatment response (Hale et al, 1989; Stewart et al, 1988) in patients with depression. At a molecular level, transporter oligomerization (Seidel et al, 2005; Sitte et al, 2004) and transmembrane ion and voltage gradients (Hilber et al, 2005) have been shown to modulate 5-HTT efficiency. In addition, regulation of 5-HTT-conducting states depends on the interaction with an associated protein, syntaxin 1A (Quick, 2003). Carneiro and Blakely (2006) have noted changes in catalytic rates of 5-HTT following changes in 5-HTT association to another protein, Hic-5. It is left to future studies to investigate the relevance of these interactions for the pathogenesis of depression in SAD.
In line with the differential effects that associated proteins exert on the functional states of 5-HTT, it has been appreciated in recent years that efflux and influx appear to be distinct and separate functions of monoamine transporters: especially, regulation of efflux is specifically influenced by phosphorylation of the N terminus of biogenic amine transporters (Fog et al, 2006; Khoshbouei et al, 2004; Seidel et al, 2005). Moreover, mechanisms of catalytic activation and inactivation have recently been proposed for 5-HTT in platelets; these involve pathways relying on the activity of protein kinases C and G, p38 mitogen-activated protein kinase (MAPK), and protein phosphatase type 2A (Jayanthi et al, 2005; Carneiro and Blakely, 2006).
Function and surface expression of the closely related norepinephrine and dopamine transporters are influenced by partly pathogenic mutations (Hahn et al, 2005, 2003; Nass et al, 2005; Shannon et al, 2000). Sequence analysis of the human 5-HTT gene in recent years has revealed over 10 variants. One of these variants is a functional mutant (I425V) that leads to increased Vmax but little change in substrate affinity (Kilic et al, 2003). In addition, a Gly56Ala substitution was found to be associated with altered catalytic activation in response protein kinase G/p38 MAPK activators (Prasad et al, 2005). It is left to future studies to determine whether some of these mutations play a role in altering 5-HTT function in SAD.
Our results bear notable similarity with results obtained in provocation studies in patients with SAD using mCPP (Garcia-Borreguero et al, 1995; Schwartz et al, 1997), an agent commonly referred to as ‘nonspecific serotonin agonist’. These studies show enhanced hormonal and behavioral response to mCPP in depressed patients with SAD and normalization after BLT. Usually, these results are interpreted as an indication of compensatory upregulation in postsynaptic serotonin receptor function during depression and subsequent receptor normalization after BLT. However, besides being an agonist at serotonin receptors, mCPP—similar to TYR—elicits a nonexocytotic, 5-HTT-mediated exchange diffusion process (Gobbi et al, 2002; Rothman and Baumann, 2002). Hence, the enhanced mCPP response in SAD and its normalization after BLT could in part also be secondary to enhanced CNS 5-HTT function normalizing after successful BLT.
According to our data, seasonal changes and alterations in platelet 5-HTT function during depression in SAD are mediated by changes in 5-HTT transport efficiency rather than changes in the amount of 5-HTT protein. This is in contrast to two earlier studies in smaller samples who report either decreased (Stain-Malmgren et al, 1998) or increased (Smedh et al, 1999) Bmax values during depression in SAD, and an increase (Stain-Malmgren et al, 1998) or decrease (Smedh et al, 1999) after BLT. In line with Ozaki et al (1994), we measured similar Bmax values in patients and control subjects. Moreover, Bmax remained stable over all three time points (see also Swiecicki et al, 2005). However, since all participants underwent BLT, and since measurements were limited to three time points, our study is not readily suited to assess naturally occurring circannual changes in 5-HTT Bmax.
Using [123I]β-CIT and SPECT, we have previously shown decreased midbrain 5-HTT availability in depressed patients with SAD (Willeit et al, 2000). It is unclear whether 5-HTT Bmax data acquired in platelets can be expected to reflect the situation in the CNS (Malison et al, 1998; Rausch et al, 2005). Several additional regulatory processes, such as degradation or pruning of new synapses and de novo synthesis of 5-HTTs may contribute to the regulation of 5-HTT quantity in the CNS. In contrast to neurons, platelets are not equipped with protein synthesis machinery and are therefore unlikely to be capable of an adaptive response with regard to 5-HTT quantity. On the contrary, circulating humoral factors mediating an adaptive (or maladaptive) response in platelet 5-HTT function may at the same time be active in the CNS, where enhanced 5-HTT function would possibly lead to decreased synaptic serotonin levels. Platelet 5-HTT Vmax—in contrast to Bmax—has recently been shown to correlate with CNS 5-HTT Vmax, provided that factors such as time of the day and gender are taken into account (Rausch et al, 2005). Matching procedures and blood withdrawals standardized for time of the day control these factors in the present study.
CONCLUSION
Reduced synaptic serotonin concentrations are a major tenet of the monoamine hypothesis of depression. Enhanced 5-HTT function as suggested by the present data may be an important mechanism contributing to low synaptic serotonin levels during depression, since it presumably leads to enhanced synaptic serotonin clearance. This mechanism is compatible with the known antidepressant effects of 5-HTT blockade in seasonal (Lam et al, 2006; Moscovitch et al, 2004) and nonseasonal (Axelrod and Inscoe, 1963) depression. Our data suggest that changes in 5-HTT efficiency are a meaningful and naturally occurring physiological process that is altered in patients with SAD, and they show that changes in 5-HTT efficiency occur parallel to changes in depressive symptoms. In view of the limitations of current techniques for the investigation of central nervous 5-HTT function, our data demonstrate the value of studying platelet 5-HTTs, and they underscore the importance of identifying mechanisms that control 5-HTT transport efficiency in humans.
References
Avery DH (1998). A turning point for seasonal affective disorder and light therapy research? Arch Gen Psychiatry 55: 863–864.
Axelrod J, Inscoe JK (1963). The uptake and binding of circulating serotonin and the effect of drugs. J Pharmacol Exp Ther 141: 161–165.
Carneiro AM, Blakely RD (2006). Serotonin-, protein kinase C-, and Hic-5-associated redistribution of the platelet serotonin transporter. J Biol Chem 281: 24769–24780.
Cesura AM, Ritter A, Picotti GB, Da Prada M (1987). Uptake, release, and subcellular localization of 1-methyl-4-phenylpyridinium in blood platelets. J Neurochem 49: 138–145.
Chang AS, Chang SM (1999). Nongenomic steroidal modulation of high-affinity serotonin transport. Biochim Biophys Acta 1417: 157–166.
Cook Jr EH, Courchesne R, Lord C, Cox NJ, Yan S, Lincoln A et al (1997). Evidence of linkage between the serotonin transporter and autistic disorder. Mol Psychiatry 2: 247–250.
Delgado PL, Price LH, Miller HL, Salomon RM, Licinio J, Krystal JH et al (1991). Rapid serotonin depletion as a provocative challenge test for patients with major depression: relevance to antidepressant action and the neurobiology of depression. Psychopharmacol Bull 27: 321–330.
D'Hondt P, Maes M, Leysen JE, Gommeren W, Scharpe S, Cosyns P (1994). Binding of [3H]paroxetine to platelets of depressed patients: seasonal differences and effects of diagnostic classification. J Affect Disord 32: 27–35.
DSM-IV (1994). Diagnostic and Statistical Manual of Mental Disorders 4th edn. American Psychiatric Association: Washington, DC.
Eastman CI, Young MA, Fogg LF, Liu L, Meaden PM (1998). Bright light treatment of winter depression: a placebo-controlled trial. Arch Gen Psychiatry 55: 883–889.
First M, Gibbon M, Spitzer R, Williams J (1996). Structured Clinical Interview for DSM-IV Axis I Disorders: Nonpatient Edition (SCID-I/NP). Biometrics Research Department, New York State Psychiatric Institute: New York, NY.
Fischer JF, Cho AKY (1979). Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J Pharmacol Exp Ther 208: 203–209.
Fog JU, Khoshbouei H, Holy M, Owens WA, Vaegter CB, Sen N et al (2006). Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron 51: 417–429.
Franke L, Schewe HJ, Muller B, Campman V, Kitzrow W, Uebelhack R et al (2000). Serotonergic platelet variables in unmedicated patients suffering from major depression and healthy subjects: relationship between 5HT content and 5HT uptake. Life Sci 67: 301–305.
Franke L, Schewe HJ, Uebelhack R, Muller-Oerlinghausen B (2003). High platelet-serotonin uptake activity is associated with a rapid response in depressed patients treated with amitriptyline. Neurosci Lett 345: 105–108.
Garcia-Borreguero D, Jacobsen FM, Murphy DL, Joseph-Vanderpool JR, Chiara A, Rosenthal NE (1995). Hormonal responses to the administration of m-chlorophenylpiperazine in patients with seasonal affective disorder and controls. Biol Psychiatry 37: 740–749.
Gobbi M, Moia M, Pirona L, Ceglia I, Reyes-Parada M, Scorza C et al (2002). p-Methylthioamphetamine and 1-(m-chlorophenyl)piperazine, two non-neurotoxic 5-HT releasers in vivo, differ from neurotoxic amphetamine derivatives in their mode of action at 5-HT nerve endings in vitro. J Neurochem 82: 1435–1443.
Greenberg BD, Tolliver TJ, Huang SJ, Li Q, Bengel D, Murphy DL (1999). Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet 88: 83–87.
Gu H, Wall SC, Rudnick G (1994). Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem 269: 7124–7130.
Hahn MK, Mazei-Robison MS, Blakely RD (2005). Single nucleotide polymorphisms in the human norepinephrine transporter gene affect expression, trafficking, antidepressant interaction, and protein kinase C regulation. Mol Pharmacol 68: 457–466.
Hahn MK, Robertson D, Blakely RD (2003). A mutation in the human norepinephrine transporter gene (SLC6A2) associated with orthostatic intolerance disrupts surface expression of mutant and wild-type transporters. J Neurosci 23: 4470–4478.
Hale AS, Sandler M, Hannah P, Bridges PKY (1989). Tyramine conjugation test for prediction of treatment response in depressed patients. Lancet 1: 234–236.
Hamilton M (1967). Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol 6: 278–296.
Harrison WM, Cooper TB, Stewart JW, Quitkin FM, McGrath PJ, Liebowitz MR et al (1984). The tyramine challenge test as a marker for melancholia. Arch Gen Psychiatry 41: 681–685.
Heils A, Teufel A, Petri S, Stober G, Riederer P, Bengel D et al (1996). Allelic variation of human serotonin transporter gene expression. J Neurochem 66: 2621–2624.
Hilber B, Scholze P, Dorostkar MM, Sandtner W, Holy M, Boehm S et al (2005). Serotonin-transporter mediated efflux: a pharmacological analysis of amphetamines and non-amphetamines. Neuropharmacology 49: 811–819.
Javitch JA, D'Amato RJ, Strittmatter SM, Snyder SH (1985). Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci USA 82: 2173–2177.
Javors MA, Seneviratne C, Roache JD, Ait-Daoud N, Bergeson SE, Walss-Bass MC et al (2005). Platelet serotonin uptake and paroxetine binding among allelic genotypes of the serotonin transporter in alcoholics. Prog Neuropsychopharmacol Biol Psychiatry 29: 7–13.
Jayanthi LD, Samuvel DJ, Blakely RD, Ramamoorthy S (2005). Evidence for biphasic effects of protein kinase C on serotonin transporter function, endocytosis, and phosphorylation. Mol Pharmacol 67: 2077–2087.
Johansson C, Willeit M, Levitan R, Partonen T, Smedh C, Del Favero J et al (2003). The serotonin transporter promoter repeat length polymorphism, seasonal affective disorder and seasonality. Psychol Med 33: 785–792.
Kasper S (1991). Jahreszeit und Befindlichkeit in der Allgemeinbevölkerung. Eine Mehrebenenuntersuchung zur Epidemiologie, Biologie und therapeutischen Beeinflussbarkeit (Lichttherapie) saisonaler Befindlichkeitsschwankungen. In: Hippius H, Janzarik W, Müller C (eds). Monographien aus dem Gesamtgebiete der Psychiatrie. Springer Verlag: New York. pp 135–136.
Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME et al (2004). N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol 2: E78.
Kilic F, Murphy DL, Rudnick G (2003). A human serotonin transporter mutation causes constitutive activation of transport activity. Mol Pharmacol 64: 440–446.
Lam RW, Levitt AJ, Levitan RD, Enns MW, Morehouse R, Michalak EE et al (2006). The Can-SAD study: a randomized controlled trial of the effectiveness of light therapy and fluoxetine in patients with winter seasonal affective disorder. Am J Psychiatry 163: 805–812.
Lam RW, Zis AP, Grewal A, Delgado PL, Charney DS, Krystal JH (1996). Effects of rapid tryptophan depletion in patients with seasonal affective disorder in remission after light therapy. Arch Gen Psychiatry 53: 41–44.
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S et al (1996). Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 274: 1527–1531.
Lesch KP, Wolozin BL, Murphy DL, Reiderer P (1993). Primary structure of the human platelet serotonin uptake site: identity with the brain serotonin transporter. J Neurochem 60: 2319–2322.
Levitan RD, Kaplan AS, Brown GM, Vaccarino FJ, Kennedy SH, Levitt AJ et al (1998). Hormonal and subjective responses to intravenous m-chlorophenylpiperazine in women with seasonal affective disorder. Arch Gen Psychiatry 55: 244–249.
Lewy AJ, Bauer VK, Cutler NL, Sack RL, Ahmed S, Thomas KH et al (1998). Morning vs evening light treatment of patients with winter depression. Arch Gen Psychiatry 55: 890–896.
Lim JE, Papp A, Pinsonneault J, Sadee W, Saffen D (2006). Allelic expression of serotonin transporter (SERT) mRNA in human pons: lack of correlation with the polymorphism SERTLPR. Mol Psychiatry 11: 649–662.
Malison RT, Price LH, Berman R, van Dyck CH, Pelton GH, Carpenter L et al (1998). Reduced brain serotonin transporter availability in major depression as measured by [123I]-2 beta-carbomethoxy-3 beta-(4-iodophenyl)tropane and single photon emission computed tomography. Biol Psychiatry 44: 1090–1098.
Maswood S, Truitt W, Hotema M, Caldarola-Pastuszka M, Uphouse L (1999). Estrous cycle modulation of extracellular serotonin in mediobasal hypothalamus: role of the serotonin transporter and terminal autoreceptors. Brain Res 831: 146–154.
Meyer JH, Houle S, Sagrati S, Carella A, Hussey DF, Ginovart N et al (2004). Brain serotonin transporter binding potential measured with carbon 11-labeled DASB positron emission tomography: effects of major depressive episodes and severity of dysfunctional attitudes. Arch Gen Psychiatry 61: 1271–1279.
Moscovitch A, Blashko CA, Eagles JM, Darcourt G, Thompson C, Kasper S et al (2004). A placebo-controlled study of sertraline in the treatment of outpatients with seasonal affective disorder. Psychopharmacology (Berl) 171: 390–397.
Nass R, Hahn MK, Jessen T, McDonald PW, Carvelli L, Blakely RD (2005). A genetic screen in Caenorhabditis elegans for dopamine neuron insensitivity to 6-hydroxydopamine identifies dopamine transporter mutants impacting transporter biosynthesis and trafficking. J Neurochem 94: 774–785.
Neuger J, El Khoury A, Kjellman BF, Wahlund B, Aberg-Wistedt A, Stain-Malmgren R (1999). Platelet serotonin functions in untreated major depression. Psychiatry Res 85: 189–198.
Neumeister A, Praschak-Rieder N, Besselmann B, Rao ML, Gluck J, Kasper S (1997). Effects of tryptophan depletion on drug-free patients with seasonal affective disorder during a stable response to bright light therapy. Arch Gen Psychiatry 54: 133–138.
Neumeister A, Praschak-Rieder N, Hesselmann B, Vitouch O, Rauh M, Barocka A et al (1998). Effects of tryptophan depletion in fully remitted patients with seasonal affective disorder during summer. Psychol Med 28: 257–264.
Nisimura T, Seto A, Nakamura K, Miyama M, Nagao T, Tamotsu S et al (2005). Experiential effects of appetitive and nonappetitive odors on feeding behavior in the blowfly, Phormia regina: a putative role for tyramine in appetite regulation. J Neurosci 25: 7507–7516.
Nobile M, Begni B, Giorda R, Frigerio A, Marino C, Molteni M et al (1999). Effects of serotonin transporter promoter genotype on platelet serotonin transporter functionality in depressed children and adolescents. J Am Acad Child Adolesc Psychiatry 38: 1396–1402.
Owens MJ, Nemeroff CB (1994). Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem 40: 288–295.
Ozaki N, Rosenthal NE, Mazzola P, Chiueh CC, Hardin T, Garcia-Borreguero D et al (1994). Platelet [3H]paroxetine binding, 5-HT-stimulated Ca2+ response, and 5-HT content in winter seasonal affective disorder. Biol Psychiatry 36: 458–466.
Parsey RV, Hastings RS, Oquendo MA, Huang YY, Simpson N, Arcement J et al (2006). Lower serotonin transporter binding potential in the human brain during major depressive episodes. Am J Psychiatry 163: 52–58.
Partonen T, Lonnqvist J (2000). Bright light improves vitality and alleviates distress in healthy people. J Affect Disord 57: 55–61.
Patkar AA, Berrettini WH, Lundy A, Murray HW, Hill KP, Vergare MJ et al (2003). Seasonal variations in the binding of [3H]paroxetine to the platelet serotonin transporter sites in African-American cocaine-dependent patients and healthy volunteers. Hum Psychopharmacol 18: 103–111.
Pifl C, Singer EA (1999). Ion dependence of carrier-mediated release in dopamine or norepinephrine transporter-transfected cells questions the hypothesis of facilitated exchange diffusion. Mol Pharmacol 56: 1047–1054.
Prasad HC, Zhu CB, McCauley JL, Samuvel DJ, Ramamoorthy S, Shelton RC et al (2005). Human serotonin transporter variants display altered sensitivity to protein kinase G and p38 mitogen-activated protein kinase. Proc Natl Acad Sci 102: 11545–11550.
Quick MW (2003). Regulating the conducting states of a mammalian serotonin transporter. Neuron 40: 537–549.
Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS et al (1993). Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci 90: 2542–2546.
Rausch JL, Johnson ME, Li J, Hutcheson J, Carr BM, Corley KM et al (2005). Serotonin transport kinetics correlated between human platelets and brain synaptosomes. Psychopharmacology (Berl) 180: 391–398.
Rosel P, Arranz B, Vallejo J, Alvarez P, Menchon JM, Palencia T et al (1999). Altered [3H]imipramine and 5-HT2 but not [3H]paroxetine binding sites in platelets from depressed patients. J Affect Disord 52: 225–233.
Rosenthal NE, Sack DA, Gillin JC, Lewy AJ, Goodwin FK, Davenport Y et al (1984). Seasonal affective disorder. A description of the syndrome and preliminary findings with light therapy. Arch Gen Psychiatry 41: 72–80.
Rothman RB, Baumann MH (2002). Therapeutic and adverse actions of serotonin transporter substrates. Pharmacol Ther 95: 73–88.
Rudnick G, Clark J (1993). From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim Biophys Acta 1144: 249–263.
Sandler M, Carter SB, Cuthbert MF, Pare CMY (1975). Is there an increase in monoamine-oxidase activity in depressive illness? Lancet 1: 1045–1049.
Scholze P, Sitte HH, Singer EAY (2001). Substantial loss of substrate by diffusion during uptake in HEK-293 cells expressing neurotransmitter transporters. Neurosci Lett 309: 173–176.
Scholze P, Zwach J, Kattinger A, Pifl C, Singer EA, Sitte HH (2000). Transporter-mediated release: a superfusion study on human embryonic kidney cells stably expressing the human serotonin transporter. J Pharmacol Exp Ther 293: 870–878.
Schuemann HJ (1960). On the liberation of pyrocatecholamines by tyramine. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol 238: 41–43.
Schwartz PJ, Murphy DL, Wehr TA, Garcia-Borreguero D, Oren DA, Moul DE et al (1997). Effects of meta-chlorophenylpiperazine infusions in patients with seasonal affective disorder and healthy control subjects. Diurnal responses and nocturnal regulatory mechanisms. Arch Gen Psychiatry 54: 375–385.
Seidel S, Singer EA, Just H, Farhan H, Scholze P, Kudlacek O et al (2005). Amphetamines take two to tango: an oligomer-based counter-transport model of neurotransmitter transport explores the amphetamine action. Mol Pharmacol 67: 140–151.
Shannon JR, Flattem NL, Jordan J, Jacob G, Black BK, Biaggioni I et al (2000). Orthostatic intolerance and tachycardia associated with norepinephrine-transporter deficiency. N Engl J Med 342: 541–549.
Sitte HH, Farhan H, Javitch JA (2004). Sodium-dependent neurotransmitter transporters: oligomerization as a determinant of transporter function and trafficking. Mol Interv 4: 38–47.
Sitte HH, Hiptmair B, Zwach J, Pifl C, Singer EA, Scholze P (2001). Quantitative analysis of inward and outward transport rates in cells stably expressing the cloned human serotonin transporter: inconsistencies with the hypothesis of facilitated exchange diffusion. Mol Pharmacol 59: 1129–1137.
Smedh K, Spigset O, Allard P, Mjorndal T, Adolfsson R (1999). Platelet [3H]paroxetine and [3H]lysergic acid diethylamide binding in seasonal affective disorder and the effect of bright light therapy. Biol Psychiatry 45: 464–470.
Smith KA, Fairburn CG, Cowen PJ (1997). Relapse of depression after rapid depletion of tryptophan. Lancet 349: 915–919.
Stain-Malmgren R, Kjellman BF, Aberg-Wistedt A (1998). Platelet serotonergic functions and light therapy in seasonal affective disorder. Psychiatry Res 78: 163–172.
Staley JK, Sanacora G, Tamagnan G, Maciejewski PK, Malison RT, Berman RM et al (2006). Sex differences in diencephalon serotonin transporter availability in major depression. Biol Psychiatry 59: 40–47.
Stewart JW, Harrison W, Cooper TB, Quitkin FMY (1988). Tyramine sulfate excretion may be a better predictor of antidepressant response than monoamine oxidase activity. Psychiatry Res 25: 195–201.
Swiecicki L, Bidzinski A, Tonderska A (2005). [Platelet serotonin transport in the group of outpatients with seasonal affective disorder before and after light treatment, and in remission (in the summer)]. Psychiatr Pol 39: 459–468.
Terman M, Terman JS, Ross DC (1998). A controlled trial of timed bright light and negative air ionization for treatment of winter depression. Arch Gen Psychiatry 55: 875–882.
Weissman MM, Wickramaratne P, Adams P, Wolk S, Verdeli H, Olfson M (2000). Brief screening for family psychiatric history: the family history screen. Arch Gen Psychiatry 57: 675–682.
Willeit M, Praschak-Rieder N, Neumeister A, Pirker W, Asenbaum S, Vitouch O et al (2000). [123I]-beta-CIT SPECT imaging shows reduced brain serotonin transporter availability in drug-free depressed patients with seasonal affective disorder. Biol Psychiatry 47: 482–489.
Willeit M, Praschak-Rieder N, Neumeister A, Zill P, Leisch F, Stastny J et al (2003). A polymorphism (5-HTTLPR) in the serotonin transporter promoter gene is associated with DSM-IV depression subtypes in seasonal affective disorder. Mol Psychiatry 8: 942–946.
Williams JD, Link MJ, Rosenthal NE, Terman M (1988). Structured Interview Guide for the Hamilton Depression Rating Scale, Seasonal Affective Disorder Version (SIGH-SAD). New York Psychiatric Institute: New York.
Wirz-Justice A (1998). Beginning to see the light. Arch Gen Psychiatry 55: 861–862.
Author information
Authors and Affiliations
Corresponding authors
Additional information
DISCLOSURE/CONFLICT OF INTEREST
This work was supported by grants of the ‘Hygiene Fonds’ of the University of Vienna (to MW, HHS, EAS), the Austrian National Bank (OeNB grant 9085 to NP-R and MW), and the Austrian Science Foundation (FWF grant P17076 to HHS). Dr Kasper has received grant/research support form Eli Lilly, Lundbeck, Bristol-Myers Squibb, GlaxoSmithKline, Organon, Sepracor, and Servier; has served as a consultant or on advisory boards for AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly, Lundbeck, Pfizer, Organon, Sepracor, Janssen, and Novartis; and has served on speakers' bureaus for AstraZeneca, Eli Lilly, Lundbeck, Sepracor, and Janssen. Dr Brannath serves as a statistical consultant for Novartis and Hoffmann-La Roche. Other authors have nothing to disclose.
Rights and permissions
About this article
Cite this article
Willeit, M., Sitte, H., Thierry, N. et al. Enhanced Serotonin Transporter Function during Depression in Seasonal Affective Disorder. Neuropsychopharmacol 33, 1503–1513 (2008). https://doi.org/10.1038/sj.npp.1301560
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301560
Keywords
This article is cited by
-
Antidepressant Medication Status as a Moderator of Winter Depression Recurrence Following Cognitive-Behavioral Therapy and Light Therapy: Is There Evidence of an Iatrogenic Effect?
Cognitive Therapy and Research (2023)
-
Light rescues circadian behavior and brain dopamine abnormalities in diurnal rodents exposed to a winter-like photoperiod
Brain Structure and Function (2018)
