
Abstract
Modafinil (2-[(Diphenylmethyl) sulfinyl] acetamide, Provigil) is an FDA-approved medication with wake-promoting properties. Pre-clinical studies of modafinil suggest a complex profile of neurochemical and behavioral effects, distinct from those of amphetamine. In addition, modafinil shows initial promise for a variety of off-label indications in psychiatry, including treatment-resistant depression, attention-deficit/hyperactivity disorder, and schizophrenia. Cognitive dysfunction may be a particularly important emerging treatment target for modafinil, across these and other neuropsychiatric disorders. We aimed to comprehensively review the empirical literature on neurochemical actions of modafinil, and effects on cognition in animal models, healthy adult humans, and clinical populations. We searched PubMed with the search term ‘modafinil’ and reviewed all English-language articles for neurochemical, neurophysiological, cognitive, or information-processing experimental measures. We additionally summarized the pharmacokinetic profile of modafinil and clinical efficacy in psychiatric patients. Modafinil exhibits robust effects on catecholamines, serotonin, glutamate, gamma amino-butyric acid, orexin, and histamine systems in the brain. Many of these effects may be secondary to catecholamine effects, with some selectivity for cortical over subcortical sites of action. In addition, modafinil (at well-tolerated doses) improves function in several cognitive domains, including working memory and episodic memory, and other processes dependent on prefrontal cortex and cognitive control. These effects are observed in rodents, healthy adults, and across several psychiatric disorders. Furthermore, modafinil appears to be well-tolerated, with a low rate of adverse events and a low liability to abuse. Modafinil has a number of neurochemical actions in the brain, which may be related to primary effects on catecholaminergic systems. These effects are in general advantageous for cognitive processes. Overall, modafinil is an excellent candidate agent for remediation of cognitive dysfunction in neuropsychiatric disorders.
Similar content being viewed by others
INTRODUCTION
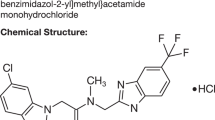
Modafinil (2-[(Diphenylmethyl) sulfinyl] acetamide; brand name Provigil in the United States) is a novel wake-promoting agent first marketed in France in the early 1990s, as a treatment for the excessive somnolence as a feature of narcolepsy. It is currently approved by the United States Food and Drug Administration as a schedule IV agent to treat excessive daytime sleepiness in narcolepsy, shift work sleep disorder, and obstructive sleep apnea/hypopnea syndrome. It has been popularly categorized as a psychostimulant due to its wake-promoting properties. However, it has shown a number of effects on physiology and behavior in both animal models and in humans, which suggest a divergent mechanism of action compared to amphetamine (described in detail below). This includes a lower liability to abuse, and a lower risk of adverse effects on organ systems such as the cardiovascular system. As a result, great interest has emerged in the possibility that modafinil may demonstrate clinical efficacy in a number of medical and psychiatric conditions currently treated with stimulants, such as various fatigue syndromes, treatment-resistant depression, and attention-deficit/hyperactivity disorder (ADHD). This interest has spawned numerous clinical trials of modafinil undertaken and reported across a range of these illnesses in recent years. These studies are summarized below, and more comprehensively reviewed elsewhere (Ballon and Feifel, 2006). The range of off-label uses for modafinil nevertheless appears to be outpacing the growth of this empirical literature, despite a lack of clear consensus about the precise neurochemical mechanism of action of this agent, inadequate clinical experience and a dearth of empirical data addressing the long-term use of this agent.
Among the various potential treatment targets for modafinil found across neurology and psychiatry, cognitive dysfunction is perhaps the target with the most critical need for truly novel pharmacotherapies, given the importance of cognition to clinical outcome in these disorders and the relative paucity of treatment options for cognition existing in the current pharmacopoeia. The emerging emphasis on cognitive dysfunction in neuropsychiatric disorders, together with the well-established effects of modafinil on arousal and activity, has inspired an emerging literature addressing the pro-cognitive effects of modafinil. These studies suggest that this agent is a promising candidate agent for cognitive dysfunction, particularly in disorders such as ADHD and schizophrenia where cognitive deficits are core, disabling features. Therefore, both the expanding list of off-label uses for modafinil and the prospects for identifying a novel pro-cognitive agent necessitate a summary and integration of the empirical literature existing to date. In this review, we briefly summarize the pharmacokinetic profile of modafinil in humans. We then outline and attempt to synthesize the complex literature addressing the neurochemical effects of modafinil, particularly as a potential treatment for cognitive dysfunction. We review the empirical literature where effects of modafinil on cognition have been tested, in animal models, healthy humans, and clinical populations. Finally, we summarize the empirical studies of clinical effects of modafinil in psychiatric disorders. Overall, this literature appears to provide a clear rationale for further investigation of the neural basis of modafinil effects on cognition, both to elaborate the role of central neurotransmitter systems in the modulation of normal cognition, and to evaluate modafinil as a candidate agent for the treatment of cognitive dysfunction.
PHARMACOKINETICS OF MODAFINIL IN HUMANS
Modafinil is a racemate, with the two enantiomers being approximately equipotent in behavioral effects in mice, but different in pharmacokinetic profile (reviewed by Robertson and Hellriegel, 2003). The R-enantiomer (armodafinil) appears to reach higher plasma concentrations than the racemic form between 6–14 h after administration, with an associated longer duration of wake-promoting activity in healthy adults (Dinges et al, 2006). Modafinil can be reliably determined in plasma and urine (Schwertner and Kong, 2005; Tseng et al, 2005), and is readily absorbed (40–65%, as measured by urinary recovery) after single (Wong et al, 1999a) or multiple oral doses (Wong et al, 1999b), reaching peak plasma concentrations 2–4 h after administration (Wong et al, 1999a). The presence of food in the gastrointestinal tract can slow the rate but does not affect the total extent of absorption. Steady-state plasma concentrations are achieved between 2 and 4 days with repeated dosing. It is highly lipophilic, and approximately 60% bound to plasma proteins, primarily albumin. Major pharmacokinetic parameters are independent of doses in the range of 200–600 mg/day (Robertson and Hellriegel, 2003). The major circulating metabolites modafinil acid and modafinil sulfone do not appear to exert any significant activity in the brain or periphery (Robertson and Hellriegel, 2003). The elimination half-life is approximately 12–15 h (Wong et al, 1999a), and single daily dosing is adequate and common in clinical practice. Elimination occurs primarily in the liver, via amide hydrolysis and a lesser component by cytochrome P450-mediated oxidation. Excretion occurs in the urine, with less than 10% of the oral dose excreted as the unchanged drug. Elimination is slowed in the elderly or in individuals with hepatic or renal impairment (Wong et al, 1999a, 1999b). Some drug–drug interactions are apparent with modafinil. In vitro, modafinil exerts a reversible inhibition of CYP2C19 (in human liver microsomes), and a smaller but concentration-dependent induction of CYP 1A2, 2B6, and 3A4, and suppression of 2C9 activity, in primary cultures of human hepatocytes (Robertson et al, 2000; Wong et al, 1999b). The 2C9 suppression observed in vitro is much less apparent in vivo. The modafinil metabolite modafinil sulfone also inhibits 2C19 with a comparable Ki. The inhibition of 2C19 may be significant for those minority of patients who are 2D6-deficient and taking concurrent medications that are substrates for 2D6 with ancillary metabolic degradation via 2C19 (eg, fluoxetine, clomipramine). Clinical studies have found significant interactions of modafinil with ethinylestradiol and triazolam (through CYP3A4 induction in the gastrointestinal system) (Robertson et al, 2002b), although not with methylphenidate (Hellriegel et al, 2001; Wong et al, 1998a), dextroamphetamine (Hellriegel et al, 2002; Wong et al, 1998b) or warfarin (Robertson et al, 2002a).
NEUROCHEMICAL EFFECTS OF MODAFINIL
Modafinil Effects on Catecholamine Systems
The empirical literature addressing modafinil effects on central neurotransmitter systems is summarized in Table 1. Modafinil is structurally unrelated to amphetamine and has a differing profile of pharmacological and behavioral effects (Table 2). An early study found modafinil to exhibit only a modest affinity for the DA transporter (DAT) (IC50=3.19 μM) in a rodent brain preparation, and no apparent specific binding to a range of other monoamine or neuropeptide receptors or transporters, nerve membrane ion channels, nor direct effects on second messenger systems in the brain (Mignot et al, 1994). However, a recent positron emission tomography (PET) study of rhesus monkeys found significant binding of the DAT (using [11C]CFT) in the striatum (54% occupancy at 8 mg/kg) and norepinephrine (NE) transporter (NET) (using [11C]MeNER) in the thalamus (44% occupancy at 8 mg/kg) (Madras et al, 2006). In addition, using in vitro human monoamine transporter preparations, binding to DAT and NET was confirmed with IC50 <10 μM (and IC50 >500 μM for the 5HT transporter). In this study, the in vitro potency of modafinil in binding DAT and NET was low relative to methylphenidate, buproprion, or benztropine; however, modafinil showed DAT occupancy by PET that was comparable to methylphenidate at clinically relevant doses. In addition, the doses used to detect DAT binding were 2–8 times lower than that which promotes wakefulness in monkeys (Hermant et al, 1991). Furthermore, whereas modafinil 10 μg did not exhibit direct binding to the trace amine receptor 1 (TA1) in vitro, it did augment the stimulation of TA1 by phenylethylamine in cells expressing DAT and NET. There is recent evidence for modulatory interactions between the TA1 receptor and both DA neuron activity in rats (Geracitano et al, 2004) and DAT activity in primates (Miller et al, 2005; Xie and Miller, 2007; Xie et al, 2007), and it is possible that TA1 receptor activity mediates some of the interactions of modafinil with DA neurons.
Other studies have reported evidence suggesting that modafinil has a mixed profile of effects on central DA systems, and lacks many neurochemical and behavioral effects observed with amphetamine administration. For instance, in contrast to amphetamine, modafinil does not affect the spontaneous release of DA from mouse striatal synaptosomes (Simon et al, 1995) or turnover of DA in the mouse caudate nucleus in vivo (De Sereville et al, 1994); it shows negligible effects on cerebral cortical blood flow (Florence et al, 2000), and different patterns of metabolic activation (Engber et al, 1998a) and c-Fos induction compared to amphetamine (Engber et al, 1998b; Lin et al, 1996); it does not produce behavioral stereotypies (Duteil et al, 1990; Simon et al, 1995) or rebound hypersomnia (Edgar and Seidel, 1997; Lin et al, 2000; Touret et al, 1995; Willie et al, 2005; Wisor et al, 2006); it does not significantly alter behavior in the elevated-plus maze (Simon et al, 1994); its effect on activity shows stress-induced subsensitivity (which is prevented by corticosterone or dexamethasone pre-treatment) (Stone et al, 2002b); pre-treatment with the tyrosine hydroxylase inhibitor α-methyl-para-tyrosine has minimal effects on modafinil-induced increases in arousal in cats (Lin et al, 1992) or activity in mice (Duteil et al, 1990; Simon et al, 1995); its effects on motor inhibitory processes are insensitive to cis-flupenthixol (a D1/D2 receptor antagonist) (Eagle et al, 2007); and in healthy humans, modafinil has effects on the resting EEG that are distinct from amphetamine (Chapotot et al, 2003). Nevertheless, parenteral administration of modafinil does lead to extracellular DA levels (measured by microdialysis) that are increased significantly in the rat prefrontal cortex (PFC) (de Saint Hilaire et al, 2001), and in the caudate nucleus of narcoleptic dogs (Wisor et al, 2001), although only minimally in the rat hypothalamus (de Saint Hilaire et al, 2001). One study found significantly increased extracellular DA in the rat nucleus accumbens, in response to intracerebroventricular modafinil dose of 10 μg (Murillo-Rodriguez et al, 2007), whereas another study found only a modest increase in DA in the accumbens after intraperitoneal (i.p.) doses up to 300 mg/kg (Ferraro et al, 1997b). Interestingly, in the first study, the modafinil effect on arousal was partly attenuated by the endocannabinoid anandamide. Modafinil effects on midbrain DA neuronal activity remain inconsistently reported. An earlier study found no effects on the activity of mesencephalic DA single units in rats (Akaoka et al, 1991), whereas a recent study found that in rat brain slices, modafinil (20 μM) inhibits the activity of ventral tegmental area DA neurons, with this effect abolished by sulpiride, blunted by nomifensine and unaffected by prazosin (Korotkova et al, 2006). These latter findings are consistent with modafinil inhibition of DA reuptake, leading to increased activation of the DA cell body autoreceptor to diminish DA cell firing. However, the derived current–voltage relationships for modafinil-evoked vs nomifensine-evoked currents showed a very different reversal potential in response to these two agents, suggesting that modafinil may exert its action in this preparation at a site distinct from the DAT. Nevertheless, modafinil effects on wakefulness are abolished in DAT-knockout mice (Wisor et al, 2001), although it should be cautioned that D2 autoreceptor function also appears severely impaired in DAT-knockout mice (Jones et al, 1999). In a rodent drug discrimination paradigm, modafinil partially generalizes to a cocaine-like stimulus (Gold and Balster, 1996); in addition, modafinil effects on activity in mice are modestly attenuated by the D1 receptor antagonist SCH 23390 (Simon et al, 1995), although not by haloperidol (Duteil et al, 1990); and the low-activity catechol-O-methyl transferase genotype is associated with greater clinical response to modafinil among adults with narcolepsy (Dauvilliers et al, 2002). In a study of healthy adults, single-dose modafinil 200 mg caused a reduction in blood prolactin levels; however, unlike the D2/D3 agonist pramipexole, it had no effect on blood growth hormone or thyroid stimulating hormone in these subjects (Samuels et al, 2006). Pre-treatment with either the selective catecholamine neurotoxin 6-hydroxy-DA or prazosin also abolishes the modafinil-induced reduction in extracellular gamma amino-butyric acid (GABA) in the neocortex (see below for discussion of effects on GABA) (Tanganelli et al, 1994, 1995). There is also evidence for a neuroprotective effect of modafinil on MPTP-induced nigrostriatal DA neuronal toxicity, even with a delayed administration that renders other DAT inhibitors ineffective (Aguirre et al, 1999; Fuxe et al, 1992). Overall, these findings suggest that modafinil effects on arousal and behavioral activity are at least partly mediated by synaptic DA, but in a manner differing from that of amphetamine, and possibly favoring corticostriatal over subcortical limbic circuits.
Modafinil also has effects on the central NE system. Whereas modafinil does not affect the activity of NE single units in the locus coeruleus (LC) of anaesthetized rats (Akaoka et al, 1991), it remains unclear if this is an artifact of anesthesia (see discussion in Souliere et al, 2000). Nevertheless, modafinil elevates extracellular NE levels in PFC (along with DA) and rostromedial hypothalamus (de Saint Hilaire et al, 2001). It also potentiates the NE-induced inhibition of sleep-promoting neurons in the ventrolateral preoptic nucleus (VLPO), although it has no effect on these neurons in the absence of exogenous NE (Gallopin et al, 2004). In addition, pre-treatment with α antagonists prazosin (which acts primarily at α1, but also has a lower affinity for α2 receptors (Hieble et al, 1995)) or phenoxybenzamine diminishes modafinil-induced increases in arousal (Lin et al, 1992) and activity in rats and monkeys (Duteil et al, 1990; Hermant et al, 1991), as does terazosin pre-treatment or α1B receptor knockouts in mice (Stone et al, 2002a). However, modafinil lacks the capacity to reduce cataplexy in dogs or humans with narcolepsy (Billiard et al, 1994; Shelton et al, 1995), a feature which is similar to other DAT inhibitors, and in contrast to α1B agonists and NET inhibitors (Mignot et al, 1993; Nishino et al, 1993). In addition, pre-treatment with low doses of the α2 antagonist yohimbine potentiates modafinil-induced wakefulness (Lin et al, 1992) and activity (Duteil et al, 1990), whereas higher doses attenuate the activity increases (Duteil et al, 1979). This apparent biphasic response to yohimbine suggests that low doses may preferentially block the inhibitory terminal α2 autoreceptor to enhance NE release and thus augment post-synaptic adrenergic receptor activation by modafinil, whereas higher doses also block post-synaptic α2 receptors, attenuating modafinil effects. This phenomenon has also been demonstrated with yohimbine effects on spatial working memory in animal models (Arnsten and Cai, 1993). These findings make it likely that post-synaptic α2 receptors mediate some of the behavioral effects of modafinil. Importantly, modafinil also augments pupillary dilation parameters (Hou et al, 2005) in a manner consistent with LC phasic responses to task-relevant events (Beatty, 1982a, 1982b; Richer and Beatty, 1987), suggesting the potential for LC/NE system effects in optimizing cognitive task performance, as described in the Aston-Jones and Cohen model outlined below (Aston-Jones and Cohen, 2005). Modest attenuation of modafinil-induced arousal and activity has also been observed after pre-treatment with the β-blocker propranolol (Duteil et al, 1990; Lin et al, 1992). Interestingly, pre-treatment with the NE-selective neurotoxin DSP-4 (which leaves DA neurons intact) does not affect modafinil-induced wakefulness, yet in these NE-lesioned mice both terazosin and the DA autoreceptor agonist quinpirole blunt the modafinil effects (Wisor and Eriksson, 2005).
Taken together, these varied findings suggest that modafinil may potentiate both DA and NE neurotransmission. It appears likely that the elevations in extracellular NE observed after modafinil are responsible for the majority of the adrenergic receptor-mediated effects, which may involve both α2 and α1 receptors. D1 and D2 receptors probably also mediate modafinil effects on cognition and behavior. In addition, however, Wisor and Eriksson (2005) have proposed that the elevated synaptic DA resulting from DAT inhibition may lead to DA activation of adrenergic receptors. Despite the common conception that DAT is strictly localized to the striatum (and absent in the frontal cortex), rodents exhibit significant levels of DAT binding in the anterior cingulate, prelimbic, and rostral areas of frontal cortex (Sesack et al, 1998; Tassin et al, 1978). In post-mortem human brains, DAT is found not only in the striatum, but also throughout the neocortex, including the PFC, albeit at relatively lower concentrations (Ciliax et al, 1999). In addition, there is indirect evidence of anatomic and functional convergence of DA and NE systems. For instance, DA and NE share a similar pattern of innervation of the medial PFC in the nonhuman primate (Lewis and Morrison, 1989). There is also indirect evidence that DA can be released from NE neurons in the medial PFC, as there are concomitant elevations of both DA and NE in the medial PFC (as well as occipital cortex) upon LC activation by either direct electrical stimulation or local infusion of α2 receptor antagonists, whereas both DA and NE are reduced in both cortical areas by local or systemic clonidine (Devoto et al, 2001, 2003, 2004a, 2004b; Kawahara et al, 2001). There remains the possibility that the enhanced DA results from competition with NE for binding to the NET, which plays an important role in terminating DA action in the cortex (Carboni et al, 1990; Moron et al, 2002). However, recent evidence suggests that the DA and NE increases in the medial PFC upon LC stimulation are somewhat independent of each other (Devoto et al, 2005). Furthermore, a subset of medial PFC neurons are responsive to both neurotransmitters (Bunney and Aghajanian, 1976). DA has an affinity for cloned mouse α1B receptors, which is on the same order of magnitude as NE (Zhang et al, 2004), and DA can activate adrenergic receptors in various brain regions (Cornil et al, 2002; Crochet and Sakai, 2003; Malenka and Nicoll, 1986). Whereas this evidence is indirect, this suggests a mechanism whereby the modafinil inhibition of DAT inhibition may be related to adrenergic receptor-mediated behavioral effects.
Modafinil Effects on GABA, Glutamate, and Serotonin Systems
Modafinil also has consistent effects on central glutamate and GABA neurotransmitter systems. It increases extracellular glutamate in the thalamus, and at higher doses, in the hippocampus (Ferraro et al, 1997a) and striatum (Ferraro et al, 1998). It also increases glutamate in the medial preoptic area and posterior hypothalamus, effects which are attenuated by the GABAA antagonist bicuculline in a dose-dependent manner (Ferraro et al, 1999). These regional glutamate effects occur at ascending doses in this order: thalamus=hypothalamus<striatum=hippocampus. Glutamate levels in the globus pallidus and substantia nigra are unchanged after the highest doses administered (Ferraro et al, 1998). The effects on glutamate may interact with adrenergic mechanisms, as NE facilitates the synaptic release of glutamate onto medial PFC pyramidal cells, an effect blocked by prazosin but not by yohimbine (Marek and Aghajanian, 1999). These glutamatergic effects do not appear to be due to effects on reuptake (Ferraro et al, 1999) or synthesis of glutamate (Perez de la Mora et al, 1999). However, there is evidence that modafinil causes increases in the cerebral glutamate–glutamine pool (along with elevations in aspartate and creatine–phosphocreatine, although not in N-acetyl aspartate or taurine), as measured by 2D COSY 1H-NMR (Pierard et al, 1995). This increase in the glutamate–glutamine pool may result from increased glutamine synthetase activity, as the mRNA content of this enzyme is increased after modafinil (Touret et al, 1994).
Modafinil also causes a dose-dependent decrease in GABA in the cortex (Tanganelli et al, 1994, 1992, 1995), the medial preoptic area and posterior hypothalamus (Ferraro et al, 1999, 1996), striatum, and globus pallidus (Ferraro et al, 1998), and at higher doses, in the hippocampus (Ferraro et al, 1997a), thalamus (Ferraro et al, 1997a), substantia nigra (Ferraro et al, 1998), and nucleus accumbens (Ferraro et al, 1997b). These regional GABA effects occur at ascending doses in this order: cortex<striatum/pallidum=hypothalamus<thalamus=hippocampus=substantia nigra=nucleus accumbens. In addition, in comparison to a single parenteral dose of modafinil, a 7-day course of parenteral administration leads to reductions of cortical GABA that are equal in magnitude but shorter-lasting (Tanganelli et al, 1994). Modafinil does not appear to directly affect the synthesis (Perez de la Mora et al, 1999; Tanganelli et al, 1995), basal or K+-evoked release, or uptake of GABA (Antonelli et al, 1998; Tanganelli et al, 1995). Interestingly, modafinil does prevent the effect of glutamate cytotoxicity on reduction of GABA release from cultured cortical neurons (Antonelli et al, 1998).
The effects on extracellular GABA appear to be mediated by modafinil effects on other neurotransmitter systems. Cortical GABA effects require intact catecholamine neurons, as pre-treatment with 6-hydroxy DA abolishes modafinil-induced reductions in GABA (Tanganelli et al, 1994), as does prazosin (Tanganelli et al, 1995). Modafinil effects on GABA are also influenced by the serotonin system (5HT). Modafinil-induced reductions in GABA are abolished in the cortex by pre-treatment with 5HT2 receptor antagonists methysergide or ketanserin (Tanganelli et al, 1992), and in the hypothalamus by the 5HT3 antagonist MDL72222 (which alone has no effect on GABA levels) (Ferraro et al, 1996). In addition, the 5HT-selective neurotoxin 5,7-dihydroxytryptamine reverses modafinil-induced reductions in cortical GABA (Tanganelli et al, 1995). Modafinil causes elevations in extracellular 5HT that are significant and dose-dependent in the frontal cortex, central nucleus of the amygdala, and dorsal raphe nucleus, but minimal in the hypothalamus (de Saint Hilaire et al, 2001; Ferraro et al, 2000, 2002). In addition, modafinil and the 5HT reuptake inhibitors fluoxetine, paroxetine, and imipramine mutually enhance the effect of each other on elevations in cortical 5HT (Ferraro et al, 2000, 2005). These studies all used microdialysis to measure extracellular 5HT and GABA. In addition, in frontal cortical slices, modafinil increases electrically evoked (but not spontaneous) 5HT efflux in a concentration-dependent manner (Ferraro et al, 2000, 2001), whereas neither type of 5HT efflux is affected by modafinil in cortical synaptosomes, in contrast to fenfluramine, which enhances both types of 5HT efflux in both cortical preparations (Ferraro et al, 2000, 2001). Taken together, this literature suggests that modafinil effects on GABA are at least partly mediated by 5HT, which do not involve direct effects on synthesis or vesicular storage of 5HT. Given that α receptors are found in high concentrations in the dorsal raphe nucleus and exert a tonic excitatory influence on raphe 5HT cell bodies (Millan et al, 2000), the modafinil effects on GABA may be mediated by adrenergic effects on 5HT activity.
Modafinil Effects on Orexin, Histamine, and Acetylcholine Systems
The clinical efficacy of modafinil in narcolepsy, a condition characterized by a severe deficiency of orexin (hypocretin) in the brain (Nishino, 2003), suggests that modafinil may have clinically relevant effects on this neurochemical system. Modafinil does increase Fos-immunoreactivity in identified orexin cells in the perifornical area of mice and rats (Chemelli et al, 1999; Scammell et al, 2000). However, modafinil induces wakefulness more potently in orexin-knockout mice than in wild-type mice, with similar patterns of Fos-immunoreactivity (Willie et al, 2005). In addition, modafinil does not bind the orexin 1 receptor (Wieland et al, 2002) and retains effects on both extracellular striatal DA and wake-promoting activity in orexin 2 receptor-deficient narcoleptic dogs (Wisor et al, 2001). Therefore, modafinil effects on arousal do not appear to be mediated through the orexin system, and the precise role of orexin in the cognitive and clinical effects of modafinil remains unknown. Modafinil also activates Fos in the tuberomammillary nucleus (TMN), which contains wake-promoting histaminergic (HA) neurons (Scammell et al, 2000), and both i.p. and intracerebroventricular modafinil elevates extracellular HA in the anterior hypothalamus (Ishizuka et al, 2003). However, direct injection of modafinil into the TMN does not affect HA release (Ishizuka et al, 2003). In addition, whereas HA neurons of the hypothalamus project widely throughout the brain (as do orexinergic neurons), they also receive significant innervation from brainstem serotonergic and catecholamine nuclei (primarily outside the LC and VTA), and the inhibition of HA neurons in the TMN during sleep is mainly due to GABAergic innervation from the VLPO (Haas and Panula, 2003). Interestingly, despite the close interaction between central HA and acetylcholine systems (Blandina et al, 2004), modafinil does not appear to affect extracellular acetylcholine in the cortex (Tanganelli et al, 1992) and does not reverse the scopolamine-induced increase in omission errors on the 5-choice serial reaction time (RT) test, in contrast to physostigmine (Waters et al, 2005). Given the multiple effects on catecholamines, 5HT and GABA described above for modafinil, it appears likely that modafinil effects on HA are mediated by one or more of these neurotransmitter systems. Nevertheless, a role for HA in a range of learning and memory paradigms is now established, with apparent opposing effects of H1 and H3 receptor activation, both of which may exert cognitive effects in interaction with cortical acetylcholine (Passani et al, 2000). It is intriguing to consider that some of the cognitive effects of modafinil may be mediated by enhancement of cortical HA effects.
Summary of Neurochemical Effects of Modafinil
In summary, modafinil is a psychostimulant that differs from amphetamine in structure, neurochemical profile, and behavioral effects. To date, the only central neurotransmitter elements to which modafinil has been demonstrated to directly bind are the DAT and NET, which it inhibits at modest potency. However, at doses used in clinical settings, modafinil may exert a significant inhibition of both catecholamine transporters. In addition, modafinil administration leads to significantly elevated extracellular DA, NE, 5HT, glutamate, and HA levels, and decreased GABA levels. These effects are particularly prominent in the neocortex, and generally less potent or minimal in various subcortical areas. The effects on DA and NE appear to be primary; effects on 5HT, GABA, glutamate, orexin, and HA may be secondary to catecholamine effects. The arousal and activity-promoting effects of modafinil are largely a function of activity in catecholamine systems, with α and β adrenergic receptors implicated and DA receptors also implicated but yet to be fully studied. Both the elevations in extracellular monoamines (including 5HT) measured by microdialysis and the effects on waking and activity mediated by catecholamines are generally observed with parenteral doses of 100 mg/kg or less. In contrast, the effects on extracellular glutamate and GABA (with the exception in the hypothalamus) generally require higher doses. Taken together, these sources of evidence suggest that the cognitive and behavioral effects seen in clinical use of modafinil are likely to be a function primarily of changes in monoamine activity rather than glutamate or GABA.
EFFECTS ON COGNITION AND NEUROBIOLOGICAL MEASURES OF INFORMATION PROCESSING
Modafinil Effects in Animal Models of Cognition
A number of studies of cognition in animal models have indicated the efficacy of modafinil for cognition (Table 3). Pre-treatment with modafinil is associated with a dose- and delay-dependent enhancement of working memory performance on a sequential alternation task in mice, without affecting exploratory or anxiety-related activity (Beracochea et al, 2001). Modafinil also dose-dependently improves the rate of spontaneous alternation as a measure of working memory performance in mice (Pierard et al, 2006). Interestingly, the optimal dose for enhancing working memory under stress conditions (immobilization or light exposure) was lower (8 mg/kg) than that under non-stress conditions (16 mg/kg); and at these doses, plasma corticosterone levels were lowered with stress (and inversely correlated with working memory performance), yet were elevated in the absence of stress. In another study of working memory, modafinil enhanced performance of rats on a delayed nonmatching to position task, which was not accounted for by the increased activity seen in the animals at the higher doses (Ward et al, 2004). It also dose-dependently improves performance of mice on a serial reversal discrimination task (Beracochea et al, 2003). This task requires mice to use current cues to rapidly adopt a context-appropriate strategy to make correct responses, and this learning curve is sensitive to damage to either the anterior cingulate (but not posterior cingulate) cortex or the mediodorsal nucleus of the thalamus (Krazem et al, 1995; Meunier et al, 1991). The anterior cingulate cortex is also an area which shows c-Fos activation after modafinil (Scammell et al, 2000). Interestingly, daily administration of modafinil (at the same dose) during learning acquisition of this task is associated with a more rapid and higher level of learning than after a single dose, whereas showing no effect on intersession perseveration or general alternation ability (Beracochea et al, 2002). This suggests a specific enhancement of the adoption of a context-dependent strategy, and also suggests that this effect is positively related to duration of treatment. Another study tested the effects on visual discrimination and visual sustained attention of oral doses of modafinil, from 8 to 64 mg/kg, administered to middle-aged female rats (Morgan et al, 2007). These investigators found no modafinil effects on visual discrimination learning, but did observe a dose- and delay-dependent effect on sustained attention, manifest as increased accuracy and speed and decreased premature responses. In this task, no modafinil effects were evident on omission errors, or measures of motivation or motor activity. In contrast, a study where rats performed the 5-choice serial RT task, modafinil in general did not appear to have effects on attention measures, as well as measures of sensorimotor and inhibitory processes (Waters et al, 2005). However, a recent report of modafinil effects on the Stop-Signal Reaction Time (SSRT) task may resolve these discrepant findings (Eagle et al, 2007). In this study, modafinil significantly decreased (ie, improved) SSRT only in those rats who exhibited relatively longer (ie, impaired) SSRT at baseline. This effect was apparent at 10 and 30 mg/kg i.p. but not at 3 mg/kg. In addition, no effects of modafinil were found on go-trial RT, and at the highest modafinil dose tested (100 mg/kg), modafinil was associated with a decrement in go-trial accuracy. These findings suggest that modafinil (at doses up to 30 mg/kg) affects the speed of the stop process rather than attention or response selection; yet at higher doses, these latter processes are affected adversely. Furthermore, in this study, cis-flupenthixol (a D1/D2 receptor antagonist) was co-administered with modafinil in a second set of experiments to test the role of D1/D2 receptors in mediating modafinil effects. Here, cis-flupenthixol (at doses of 0.01 or 0.04 mg/kg i.p.) showed no effect on the modafinil-mediated decrease in SSRT, or when administered alone. Conversely, modafinil (at 10 mg/kg) failed to antagonize the cis-flupenthixol-mediated increase in go-trial RT, in contrast to methylphenidate 1 mg/kg, which did block this effect of cis-flupenthixol. These results suggest that D1 or D2 receptors do not mediate the effects of modafinil on inhibitory processes as measured in this task.
Modafinil Effects on Cognition in Healthy Non-Sleep-Deprived Adults
Modafinil appears to enhance cognitive performance in healthy adults who are not sleep-deprived (Table 3). In one randomized, placebo-controlled single-dose study of 60 adults, modafinil improved performance on digit span, visual recognition memory, spatial planning, and SSRT, suggesting improved working memory and inhibition of pre-potent responding (Turner et al, 2003). No differences were found between the 100 and 200 mg single doses. Other studies have found delay-dependent improvements in working memory, such as on maintenance and manipulation and delayed matching tasks, without a speed-accuracy trade-off, or effects on attention tasks (Muller et al, 2004); and on vigilance, but not perceptual, arithmetic, or reasoning performance (Baranski et al, 2004). A different research group has found a single dose of modafinil 100 mg to improve performance on digit span and a sustained attention task (Randall et al, 2005b), yet failed to find significant improvement on a range of other cognitive tests with single doses of 100 or 200 mg modafinil in this and other studies. However, this group has studied university students who appear to have a high IQ (average of 115 in one study), with likely general ceiling effects on performance (Randall et al, 2003, 2005a). Indeed, a retrospective analysis of the studies of students found modafinil effects on cognition only for a subgroup with relatively lower IQ (Randall et al, 2005a). In another study (Randall et al, 2004), a group of relatively older subjects (aged 50–67) was studied, which may include individuals with age-related decline that involves neurochemical systems unaffected by modafinil, such as acetylcholine (Tanganelli et al, 1992).
Modafinil Effects on Cognition in Healthy Sleep-Deprived Adults
Several studies of modafinil effects on cognition in healthy adults undergoing sleep deprivation or simulated night shifts have been reported (see (Wesensten, 2006) for review). One study of adults with 85 h of sleep deprivation found single-dose modafinil 400 mg to reduce errors on the Wisconsin Card Sort Test (WCST) and interference on the Stroop (compared to placebo), and comparable to 600 mg caffeine and 20 mg amphetamine (Wesensten et al, 2005). Another study from this research group found minimal effects of modafinil single-dose (100, 200, or 400 mg) on measures of RT or arithmetic performance (Wesensten et al, 2002), suggesting that the improvement in executive functions found in their other study was not merely due to enhanced speed of response. A study of 41 military recruits, who received modafinil 300 mg, d-amphetamine 20 mg, or placebo on three separate occasions of 64 h of continuous work, found both medication treatment groups to perform better than the placebo group on short-term memory, logical reasoning, and RT measures (Pigeau et al, 1995). A double-blind, placebo-controlled study of emergency department physicians participating after an overnight work shift found single-dose modafinil 200 mg to improve accuracy (relative to placebo) on both AX and AY conditions of the AX-CPT task (Gill et al, 2006). One study of healthy adults undergoing simulated night-shift work found a 4-day regimen of modafinil 200 mg to reduce errors (compared to placebo) on the WCST and the Hayling Sentence Completion Test (Walsh et al, 2004), which requires cognitive control and is associated with activation of dorsolateral PFC (Nathaniel-James and Frith, 2002) and anterior cingulate cortex (Nathaniel-James et al, 1997) measured by fMRI. Another double-blind, placebo-controlled study of healthy adults undergoing simulated day- and night-shift conditions found a 3-day course of modafinil 200 or 400 mg to improve performance on divided attention, immediate recall, and a version of the digit-symbol task, relative to placebo (Hart et al, 2006). Modafinil effects were generally as strong at the 200 as the 400 mg dose, with stronger effects during the night-shift than day-shift condition. A randomized, placebo-controlled fMRI study of single-dose modafinil 200 mg after overnight sleep deprivation in eight healthy men found this treatment to improve working memory performance and associated cortical activation under intermediate working memory loads, using the N-Back (Thomas and Kwong, 2006).
Modafinil Effects on Cognition and Brain Function in Clinical Populations
A few studies of cognition and functional neuroanatomy have been conducted in patients with narcolepsy (Table 3). An fMRI study of narcolepsy patients and healthy controls found no within- or between-group differences in modafinil vs placebo effects on extent of activation across the whole brain in passive response to combined visual and auditory stimulation (Ellis et al, 1999). This suggests that modafinil does not merely cause diffuse activation across the cortex, as might result from primary effects on arousal or early sensory processes. A multicenter randomized, double-blind placebo-controlled 12-week study of armodafinil effects in 196 narcolepsy patients included one group receiving 150 mg/day, and another group receiving 250 mg/day. This study found armodafinil to be associated with several effects on cognition: on a summary RT measure from 3 RT tests, the low-dose and pooled low/high-dose groups performed significantly faster than the placebo group at the final (week 12) assessment; on a measure of overall accuracy across four episodic recall and recognition tasks, each armodafinil-treated group performed significantly better than placebo at 4 weeks with this difference maintained throughout the remainder of the study; and the high-dose and pooled-dose groups were significantly faster on an RT measure derived from the working memory and episodic recognition memory tasks (Harsh et al, 2006). Other studies have examined effects on scalp electrophysiology measures in narcolepsy patients. A 3-week treatment with modafinil 400 mg/day remediated the decrement in α-2 and β-1–3 power in a vigilance-controlled EEG (measured by low-resolution brain electromagnetic tomography, LORETA) that was observed in placebo-treated patients with narcolepsy; in this sample, modafinil treatment was also associated with decreases in power in the θ and δ bands in the resting EEG (Saletu et al, 2004). The remediating effects on α and β power were localized to several cortical regions, including frontal and cingulate cortex. In a related study, this group found that modafinil treatment of medication-free narcolepsy patients (titrated from 100–400 mg/day over 3 weeks) was associated with significantly improved performance on a test of simple arithmetic (Pauli Test) and effects on the EEG (by LORETA) similar to those found in the earlier study (Saletu et al, 2007). Furthermore, Pauli Test performance was correlated with modafinil effects on decreased θ and δ power, and these correlations were particularly localized to the frontal and anterior cingulate cortices. In addition, among narcolepsy patients, who exhibit a prolonged auditory and visual P300 latency (Sangal et al, 1999a), a relatively shorter P300 latency was associated with clinical response to modafinil (at either 200 or 400 mg/day), using a measure of daytime sleepiness (Sangal et al, 1999b). A shorter auditory P300 latency was also associated with remediation of fatigue in patients with multiple sclerosis in response to 4 weeks of modafinil 100 mg/day (Nagels et al, 2007). A study of scalp somatosensory evoked potentials in healthy adults found specific effects of modafinil 100 mg single dose on the short-latency component of high-frequency (500–700 Hz) oscillations, with a wide scalp distribution over the scalp and uniform polarity, and dipole modeling suggesting a subcortical source likely to be located in the brainstem (Della Marca et al, 2004). Whereas it is not entirely certain how to resolve this finding with the reported effects on the other EEG phenomena, it is possible that this last effect represents activation of brainstem centers with a diffuse cortical distribution, such as the monoamine nuclei, whose activity may be associated with widespread effects on other cortical electrical phenomena such as the other frequency bands. This issue may be best resolved by testing modafinil effects either in animal models, where single-unit or multiunit activity can be compared to simultaneous scalp electrical activity, or in humans with both scalp EEG and whole-brain imaging by fMRI.
Modafinil effects on cognition have been studied as well in psychiatric populations (Table 3). This includes a study of 20 patients with stable chronic schizophrenia, in a double-blind, placebo controlled, single-dose crossover study (Turner et al, 2004b). In this study, the modafinil 200 mg dose (added to concurrent atypical antipsychotic medications) was associated with significantly improved performance (relative to placebo) on digit span (forwards and backwards) and trends toward better performance on delayed visual recognition memory and a version of the Tower of London. In addition, on modafinil, these patients made fewer extradimensional shift errors on the intradimensional/extradimensional shift (ID/ED) task. In this visual discrimination learning task (developed as a WCST analog that could be performed by animals), the ED shift is a form of attentional set-shifting mediated by fronto-cortical loops that are modulated by ascending DA systems. Interestingly, ID/ED performance enhancement was not observed by the same group in similarly-designed studies of modafinil in patients with ADHD (Turner et al, 2004a) or healthy adults (Turner et al, 2003), who showed a pattern of performance improvement similar to each other (see below), but different from the patients in the schizophrenia study. This suggests a measure of specificity to patients with schizophrenia for enhancement of attentional set-shifting, a function strongly dependent in this task on lateral PFC (Dias et al, 1996). Modafinil had no effect on SSRT in these patients, which may be due to a higher dosage threshold for SSRT effects, as decreased SSRT was seen in healthy rats performing this task only at higher doses (10 and 30 mg/kg i.p., but not at 3 mg/kg, a dose very comparable to the 200 mg oral dose used with the schizophrenia patients) (Eagle et al, 2007). An open-label study of 11 chronic schizophrenia patients found add-on modafinil (titrated from 100 mg/day on days 1–14 to 200 mg/day on days 15–28) to improve performance on letter-number sequencing (Rosenthal and Bryant, 2004). In a double-blind, placebo-controlled study of 17 schizophrenia patients, modafinil 100 mg single-dose was associated with greater activation of the dorsal anterior cingulate cortex during performance of an N-back Task (Spence et al, 2005). In another double-blind, placebo-controlled fMRI study of 12 schizophrenia patients with prominent negative symptoms (a subset of the sample in Spence et al, 2005), this research group found modafinil 100 mg single-dose to be associated with increased bilateral dorsolateral PFC activity (Brodmann's areas 9 and 46) during performance of a task requiring subjects to press a button in an aperiodic manner (Hunter et al, 2006). Left BA 46 activity was significantly associated with the temporal variation in interresponse intervals (‘coefficient of variation’), the primary measure of task performance. The placebo-condition coefficient of variation was negatively associated with changes in both this behavioral measure and BA 46 neural activity, suggesting that those patients with worse baseline performance exhibited the strongest response to modafinil. This group has also reported that after a single modafinil dose of 100 mg, schizophrenia patients exhibited a significantly greater amount of behavioral activity than placebo-treated patients, measured with wrist-worn actigraphy over a 20-h period on an inpatient research unit (Farrow et al, 2006). Two studies of add-on modafinil treatment of schizophrenia patients have failed to find significant differences from placebo on behavioral cognitive measures. In the first, 20 clinically stable but moderate to severely ill (Clinical Global Impression scale (CGI) ⩾4) chronic schizophrenia patients performed the following tasks at baseline and again after 8 weeks of double-blind add-on modafinil, with doses of 100 or 200 mg/day: CPT-Identical Pairs version, Letter-Number Span, oculomotor delayed response, delayed match-to-sample, verbal (letter) fluency, and the Rey Auditory Verbal Learning Test (Sevy et al, 2005). On the CPT-IP, the effect size (Cohen's d) from baseline to week 8 within the modafinil group was approximately 0.3 for a few measures, whereas within the placebo group, it was approximately 0.1. On the Letter-Number Span, within the modafinil group, d was approximately 0.5 vs −0.5 within the placebo group. These results suggest that the small sample sizes (10 patients completing the study in each group) conferred inadequate statistical power to detect between-group differences on these measures. In addition, the modafinil group was relatively worse in performance at baseline on most of the other cognitive measures, whereas the placebo group exhibited a significant response on the clinical measures. In the second study (available only as an abstract), a total of 20 patients were enrolled, with no significant effects of 8-week modafinil 200 mg/day found on the California Verbal Learning Test, Degraded-Stimulus Continuous Performance Test, or Trails Part B (Pierre et al, 2005). The abstract does not indicate how many subjects completed the study. These studies appear to remain inconclusive regarding null findings with modafinil on cognitive dysfunction in schizophrenia and provide emphasis on the critical need for adequate statistical power in clinical trials study design.
In a study of patients with major depression (with 31 completers), modafinil improved another prefrontal-dependent measure, Stroop interference, in a 4-week open-label trial with flexible dosing between 100 and 400 mg/day added to existing antidepressant medications (DeBattista et al, 2004). In a double-blind 3-week trial comparing 400 vs 600 mg/day in 24 patients with narcolepsy, modafinil reduced errors on the WCST (Schwartz et al, 2004). The two doses were not directly compared for cognitive effects in this study.
As indicated above, modafinil effects on cognition have also been studied in ADHD. In a study of 20 adult ADHD patients, a single dose of modafinil 200 mg was associated with significant enhancements in performance on digit span, visual recognition memory, spatial planning, and SSRT, relative to placebo (Turner et al, 2004a). The patients as a group showed slowed latencies together with increased accuracy on several measures, including the Delayed Match-to-Sample, Tower of London, and visual recognition memory tasks, suggesting that modafinil effects including shifting individuals on the speed–accuracy curve to optimize performance. In contrast, a 2-week study of 22 adult ADHD patients, where the modafinil-treated group was titrated over 4–7 days to an average dose of 206.8 mg/day, and another group received amphetamine at an average dose of 21.8 mg/day, verbal (letter) fluency was improved relative to the placebo group, but no treatment effects were observed on the Stroop or Digit Span tests (Taylor and Russo, 2000). Performance on a version of the CPT (the Test of Variables of Attention, TOVA) has also been remediated in several studies of child/adolescent ADHD patients. This includes an open-label study in 11 children with ADHD, with an average dose of 195 mg/day for an average 4.6 weeks (Rugino and Copley, 2001); a follow-up study of 22 children with ADHD, using a randomized, placebo-controlled design with an average dose of 264 mg/day for an average of 6 weeks (Rugino and Samsock, 2003); and in a recent, much larger study of childhood ADHD, which included 100 completers in the modafinil-treated group and 41 completers in the placebo group, an average dose of 361 mg for an average of 31.5 days (Greenhill et al, 2006). In these two latter studies, overall TOVA performance improved in the modafinil-treated group, whereas it declined from pre-treatment baseline in the placebo group.
Summary of Effects of Modafinil on Cognition
These studies show consistent evidence for the benefits of modafinil for cognitive function. Studies in rodents indicate that modafinil can improve working memory performance in a dose- and delay-dependent manner, that the processing of contextual cues is also enhanced with modafinil, and that these effects may be augmented with sustained dosing regimens. In healthy humans (with or without undergoing sleep deprivation), working memory, recognition memory, sustained attention, and other tasks dependent on cognitive control are enhanced with modafinil. Some evidence suggests that the magnitude of modafinil effects in healthy adults may depend on underlying cognitive abilities. Among psychiatric populations, there is now consistent evidence that modafinil (in well-tolerated dosing regimens) improves attention and response inhibition in children and adolescents with ADHD; this benefit may be related to modafinil effects in modulating performance along the speed-accuracy curve for responsive individuals. Among adult psychiatric patients, there is evidence that modafinil improves several prefrontal-dependent cognitive functions in schizophrenia, major depression, and adult ADHD. Some null findings have been reported in schizophrenia; however, these studies have significant limitations evident in their design. The range of clinical samples and cognitive functions that are subject to modafinil treatment study is expected to expand in the future.
Mechanisms of Catecholamine Action in the Modulation of Cognition
The most highly elaborated model of catecholamine modulation of higher cognition has been developed for PFC dopamine in working memory, based primarily on studies of nonhuman primates. In particular, the D1 receptor in the DLPFC is critical to spatial working memory performance in monkeys (Sawaguchi and Goldman-Rakic, 1991, 1994). D1 receptors in the PFC are primarily found on the distal dendritic spines of pyramidal cells, often in conjunction with asymmetric, presumably glutamatergic synapses, and occasionally in triads which also include DA terminals (Smiley and Goldman-Rakic, 1993; Smiley et al, 1994; Williams and Goldman-Rakic, 1993). This may represent a post-synaptic site where D1 receptors can gate glutamatergic transmission, as D1 activation not only directly excites pyramidal neurons, but enhances the responsiveness of the post-synaptic NMDA receptor on those cells as well (Seamans and Yang, 2004). The facilitation of NMDA effects on intracellular calcium via calcyon–Gq interactions has been proposed as one of the most important functions of DA in the PFC, by not only by supporting persistent (delay-related) activity, but also by influencing both short and long-term plasticity, gene expression, and neuroadaptation (see discussion in Williams and Castner, 2006). A second major site in the PFC for the D1 receptor is at the glutamatergic terminals between neighboring pyramidal cells (Gao et al, 2001). At this site, D1 receptor activation leads to the attenuation of recurrent excitation within cortical microcircuitry, probably by presynaptic inhibition of glutamate release (Seamans and Yang, 2004). This may have the effect of constraining the extent of local activation during cognitive processes. A third major site of D1 receptors in PFC is on subtypes of GABAergic neurons (Muly et al, 1998; Sesack et al, 1998). This may serve to facilitate a feedforward inhibition that further restricts the extent of local circuit activity. Taken together, these three mechanisms of D1 receptor-mediated action in the PFC appear to potentiate intense focal activity, whereas dampening the responsiveness of the local surrounding circuitry that would otherwise compete with the presently active circuit (Goldman-Rakic et al, 2004). The information processing consequences of these physiological effects may be as follows: in a scenario of increased afferent glutamatergic activity, which informs the PFC of both when to initiate persistent activity and what the information content is, D1 receptor activation then adjusts the gain (ie, the strength of the representation) of the glutamate-encoded information in the PFC (Seamans and Yang, 2004). This includes a depression of background PFC activity, which serves to make the self-sustained activity robust to noise (eg, distractors) (Durstewitz and Seamans, 2002). A recently refined model of DA effects on PFC-mediated context processing, derived primarily from connectionist computational modeling studies, similarly suggests that optimal phasic DA action in PFC is required for the adequate processing of task-relevant stimuli, that is, the representation of contextual information (Braver et al, 1999), and DA serves a gating function by regulating the access of context representations into ‘active’ (eg, working) memory.
NE is implicated as well in PFC-dependent cognitive functions. For instance, α2 receptors strongly modulate working memory performance in monkeys and rodents. Importantly, there does not appear to be an inverted-U-shaped curve relating working memory performance to α2 agonist dose (Arnsten, 2004), and these effects probably occur at post-synaptic sites (Arnsten and Goldman-Rakic, 1985; Cai et al, 1993), where α2 receptors are found on asymmetric (probably excitatory) synapses on dendritic spines in the PFC of monkeys (Aoki et al, 1994, 1998). In contrast, preferential activation of the presynaptic α2 autoreceptor impairs working memory, probably by reducing the terminal release of NA with reduced post-synaptic α2 receptor activation as a result (Arnsten and Goldman-Rakic, 1985). The role of post-synaptic α2 receptor-mediated transient increases in PFC delay-related activity (Li et al, 1999; Sawaguchi, 1998), and associated mitigation of interference in task performance (Arnsten and Contant, 1992), suggest a point of convergence of the Arnsten model of adrenergic function with the Aston-Jones and Cohen (Aston-Jones and Cohen, 2005) model of phasic LC activity in optimizing task performance (see below).
In the Aston-Jones and Cohen (Aston-Jones and Cohen, 2005) model, phasic LC activity is driven by the outcome of task-related decision processes (signaled by descending projections from the ACC and orbitofrontal cortex), and subsequently adjusts the gain in target neurons via ascending projections back to PFC. During high (accurate) performance of visual target-detection tasks, monkeys exhibit LC activity characterized by moderate tonic activity and additional phasic responses that are selectively observed to targets (but not distractors) (Aston-Jones et al, 1994). The phasic activity is not related to the sensory features or a specific reward associated with the target stimuli, and is observed even if targets are presented on every trial. In contrast, no phasic response to distractors is seen even if distractors are infrequent. Moreover, in reversal tasks, LC activity quickly re-sets to the new target and is extinguished to the new distractor; this precedes behavioral reversal within a single testing session (Aston-Jones et al, 1997).
Tonic LC activity, on the other hand, is proposed to facilitate disengagement of the animal from the task, because during elevations in tonic LC activity, the animal exhibits less frequent foveation to targets, lower signal-detection performance (ie, lower d and β) (Aston-Jones et al, 1994), and more aborted trials (Aston-Jones et al, 1996, 1998). This is considered adaptive in allowing the animal to pursue alternative behaviors or cognitive processes (Aston-Jones and Cohen, 2005). An important role for α2 receptors in this model provides a link to the model described by Aston-Jones et al (1994). Administration of the α2 agonist clonidine leads to decreased tonic LC activity (mediated via LC cell-body autoreceptors), with concomitant increased phasic LC activity to targets, and improved performance by decreased false-alarm and omission errors (Aston-Jones and Cohen, 2005). This reciprocal relationship between tonic and phasic modes of LC activity may be mediated by changes in the degree of electrotonic coupling between LC cells (Aston-Jones and Cohen, 2005; Usher et al, 1999). It appears also that when levels of tonic LC activity are minimal, such as during sleep, grooming, and eating, that phasic responses are also less robust (Aston-Jones and Bloom, 1981). This suggests that, as with other catecholamine-mediated phenomena, phasic LC activity may be related to tonic activity in an inverted-U-shaped manner. For individuals with excessively-low tonic LC activity, enhancements of both tonic and phasic LC activity may possibly be elicited in concert.
One important implication of the inverse relationship between phasic activity and moderate to high levels of tonic activity is that agents with α2 agonist activity could act at two distinct sites to improve cognitive performance: (1) at the cell-body autoreceptor to adjust the balance of phasic to tonic LC activity in a manner to optimize decision-making performance; (2) at the post-synaptic α2 receptor to enhance sustained PFC activity (Arnsten, 2004).
CLINICAL EFFECTS OF MODAFINIL
Modafinil has consistently shown efficacy in measures of alertness in narcolepsy and shift-work sleep disorder. Two randomized, double-blind placebo-controlled studies (with a total of 554 patients) conducted by the US Modfinil in Narcolepsy Multicenter Study Group (1998, 2000) found significant efficacy of modafinil for subjective and objective measures of wakefulness among patients with narcolepsy. Similar results have been found in smaller double-blind, placebo-controlled studies (Billiard et al, 1994; Broughton et al, 1997). In these studies and others, open-label extensions have found modafinil to have long-term efficacy for sleepiness extending for as long as 136 weeks, and to be well-tolerated, with no evidence of significant adverse events or abuse (Besset et al, 1996; Hirshkowitz et al, 2006; Mitler et al, 2000). Modafinil has also shown efficacy for shift work sleep disorder, with a large randomized, double-blind placebo-controlled study showing improvements in sleep latency, vigilance, sleep-related function, and the rate of automobile accidents during the post-work commute (Czeisler et al, 2005). Modafinil has also been evaluated for the treatment of fatigue and sedation in a number of other neurological and medical conditions, including multiple sclerosis, idiopathic Parkinson's disease, chronic fatigue syndrome, polio, HIV infection, dementias, obstructive sleep apnea, post-anaesthetic sedation, and fibromyalgia, with generally favorable but somewhat mixed results (see comprehensive summary of these studies in Ballon and Feifel, 2006). Remarkably, despite the importance of cognitive dysfunction in a range of neurological and medical illnesses, to our knowledge there have been no reports to date of modafinil effects on cognition in these disorders.
Among studies of adult psychiatric patients using clinical outcome measures, adjunct modafinil has shown efficacy in a 4-week open-label study of 11 stable patients with chronic schizophrenia or schizoaffective disorder, with dosing at 100 or 200 mg/day (Rosenthal and Bryant, 2004). Of the patients, 82% completed the study, and a blinded clinician rated 64% of patients as clinically improved at week 4, using CGI and the Global Assessment of Function, with fatigue scores also improved. PANSS scores were unchanged, indicating that positive symptoms were not exacerbated, and no serious adverse events were detected. A randomized, placebo-controlled 8-week study of adjunct modafinil 100 or 200 mg/day in 13 schizophrenia patients (and 11 patients receiving placebo) found no changes in positive or negative symptoms (Sevy et al, 2005). Two studies of patients with major depression have been reported. In the first, a 4-week open-label adjunct modafinil (with flexible dosing from 100–400 mg/day) was associated with significant improvements in the Beck Depression Inventory, Hamilton Depression Rating Scale (Ham-D) and CGI, as well as measures of fatigue (DeBattista et al, 2004). The other study was a multicenter, randomized, placebo-controlled 8-week study of adjunct modafinil 200 mg/day (added to concurrent treatment with selective serotonin reuptake inhibitors), which found an 85% completion rate (of 311 patients who received at least one dose), and significant improvements in Ham-D, MADRS, and sleepiness ratings compared to placebo (Fava et al, 2005). Adverse events significantly associated with modafinil included nausea (9 vs 2% on placebo) and feeling jittery (4 vs 1%). In a 12-week, open-label extension study of these depressed patients, with modafinil doses titrated following the initial 8-week placebo-controlled study cited above, the initial modafinil non-responders showed a significantly greater clinical response on all measures than the initial treatment-responsive group (Thase et al, 2006). In an 8-week randomized, double-blind placebo-controlled study of 62 cocaine-dependent adults, modafinil 400 mg/day was associated with greater rates of urine samples that were negative for a cocaine metabolite, and of achievement of at least 3 weeks of complete abstinence from cocaine use (Dackis et al, 2005). Of the patients, 65% completed the study, and no serious adverse events were noted. A randomized, double-blind, placebo-controlled three-phase crossover study of 22 adults with ADHD found improvements in DSM-IV ADHD Behavior Checklist for Adults compare to placebo, for both modafinil (2 weeks after titration to mean 207 mg/day) and amphetamine (Taylor and Russo, 2000).
Among child and adolescent psychiatric disorders, modafinil has only been studied to date in ADHD. It has been found to improve parent, teacher, and clinician ratings of ADHD symptoms in open-label treatment of 11 medication-free children with an average dose and duration of 195 mg/day (range 100–400 mg) and 4.6 weeks (range 2–7 weeks) (Rugino and Copley, 2001). In a follow-up study of 22 children with ADHD, this time employing a randomized, placebo-controlled design with an average dose of 264 mg/day (range 200–300 mg) for an average of 6 weeks, they found the modafinil-treated group to exhibit significantly greater improvement than the placebo group on the Conners Rating Scales ADHD total score (Rugino and Samsock, 2003). The Modafinil ADHD Study Group has conducted several randomized, double-blind placebo-controlled studies of modafinil in children and adolescents with ADHD. In a 4-week study with 223 children (aged 6–13 years) completing the study, the group receiving 300 mg/day showed a significantly greater improvement in the teacher-rated ADHD Rating Scale-IV (ADHD-RS-IV), clinician-rated ADHD-RS-IV, and the parent-rated Conners ADHD/DSM-IV scales (Biederman et al, 2006). In a 7-week study with 190 ADHD patients (aged 6–17 years) enrolled, the modafinil-treated groups (receiving either 340 mg (n=44) or 425 mg (n=82), based on body weight) showed significantly greater improvement on the ADHD-RS-IV School and Home versions and on the CGI (Swanson et al, 2006). In a 9-week multicenter study of children and adolescents with ADHD (aged 7–17 years) that included 100 completers in the modafinil-treated group and 41 completers in the placebo group, modafinil at an average dose of 361 mg (range 170–425 mg) for an average of 31.5 days (range, 2–56 days) was associated with a significantly greater improvement in the ADHD-RS-IV School and Home versions, and on the Clinical Global Impression (CGI) scale (Greenhill et al, 2006). And in a 9-week multicenter study of children and adolescents with ADHD, the modafinil-treated group (n=164), receiving an average dose of 368.5 mg/day (range 170–425 mg) showed greater improvements in the ADHD-RS-IV School and Home versions, and on the CGI, compared to the placebo-treated group (Biederman et al, 2005). The significant group differences in ADHD-RS-IV School version were apparent in the first week of treatment and maintained throughout the treatment period.
Throughout these clinical intervention studies, modafinil has been well tolerated. Nevertheless, case reports have appeared describing significant adverse events in routine clinical use of modafinil. One case report has appeared describing exacerbation of psychosis in a 61-year-old inpatient, with schizophrenia and hypertension, after initiation of modafinil treatment (Narendran et al, 2002). This patient received a 3-week regimen of 800 mg/day added to existing treatment with clozapine 300 mg/day, lorazepam 1 mg/day, and amlodipine 40 mg/day. The patient is reported to have stabilized within 2 weeks after discontinuation of modafinil (including severity of positive psychotic symptoms) with no other medication changes, and there is no indication in the report of serious sequelae in the intervening period of worsened psychosis. Whereas other single case reports have appeared describing adverse events in the treatment of psychiatric patients such as clozapine toxicity (Dequardo, 2002), premature ventricular contractions (Oskooilar, 2005), induced mania (Vorspan et al, 2005; Wolf et al, 2006), and irritability and verbal aggression (Ranjan and Chandra, 2005), these events have not been observed at a significant rate in modafinil-treated patients compared to placebo-treated patients in the clinical trials cited above. Modafinil also appears to have a relatively low potential for abuse, which may be a function of its pharmacodynamic profile and/or its physical properties, being insoluble in water and unstable at high temperatures, which minimizes its bioavailability upon smoking or intravenous use (Jasinski, 2000; Myrick et al, 2004). In addition, a preliminary study of 12 cocaine-dependent adults suggests that modafinil (up to 800 mg as an open-label single-dose) does not exhibit interacting effects with 40 mg intravenous cocaine on hemodynamic measures (Malcolm et al, 2006).
Modafinil effects on anxiety have also been measured, in animal models and in humans. One study found that whereas amphetamine increased three measures of anxiety in mice, with increased latency of exploration of a white compartment, increased open-field thigmotaxis, and decreased time in the open arms of an elevated-plus maze, modafinil lacked these effects at doses that induce comparable effects on locomotor activity (Simon et al, 1994). A study of wake-promoting effects in monkeys reported no significant observations of anxiety responses to modafinil after single or repeated doses that increased nocturnal activity (Hermant et al, 1991). In contrast, a pharmacokinetic study of modafinil (at doses from 200–800 mg p.o. over 7 days) in healthy subjects found 21% to indicentally report subjective anxiety (although rates of self-reported anxiety among the placebo group are not reported) (Wong et al, 1999b). A study of mood and cognitive function in healthy young adults found a single 100 mg dose of modafinil to be associated with increased subjective and physical symptoms of anxiety (eg, restlessness, muscular tension, shaking) than placebo, although the higher dose (200 mg) did not show these effects (Randall et al, 2003). A study of healthy adults given modafinil 400 mg p.o. daily for 3 days found relatively decreased self-reported scores compared to placebo on the Calm scale of the Positive- and Negative-Affect Scale (Taneja et al, 2007). Interestingly, in this study, both overall positive and negative affect was relatively increased on modafinil. Among myotonic dystrophy patients, modafinil (100 mg p.o. daily for 14 days) increased self-reported scores on the tension–anxiety index of the Profile of Mood States (along with increased vigor-activity and decreased fatigue-inertia) compared to placebo (MacDonald et al, 2002). Two studies of obstructive sleep apnea patients reported on anxiety. In one, rates of anxiety were 6% on modafinil vs 1% on placebo (during the double-blind phase) and 16% after 12 weeks of open-label modafinil (200–400 mg/day) (Schwartz et al, 2003). In the second, rates of anxiety were 5.3% on armodafinil (150 and 250 mg/day) vs 2% on placebo (Roth et al, 2006). A study of 50 multiple sclerosis patients found three leading to drop out or dose reduction due to nervousness or restlessness (Zifko et al, 2002). It does appear, therefore, that modafinil (at clinically-effecive doses) is associated with increased anxiety in healthy individuals and clinical populations, although it is unclear if this is dose-related.
CONCLUSION
Modafinil is an agent with a rapidly expanding list of off-label uses in neurology, medicine, and psychiatry. It appears to have multiple effects on catecholamine systems in the brain, including DAT and NET inhibition, and elevation of extracellular catecholamines, glutamate, serotonin, and HA, activation of the orexinergic system, and decreased GABA. Alpha-adrenergic, D1 and D2 receptors in the brain mediate modafinil effects on waking and activity, and may also mediate the neurochemical effects on these other neurotransmitter systems. Modafinil is also significantly different from amphetamine in structure and profile of neurochemical and behavioral effects. Intriguing preliminary evidence suggests that modafinil may be relatively selective for cortical over subcortical effects. In the clinical setting, modafinil shows efficacy in a number of neurological and psychiatric illnesses, with a significantly improved side-effect profile compared to amphetamine, including a relatively low liability to abuse. Equally important, there is now increasing evidence that modafinil can improve cognitive function, particularly working memory, episodic memory, and processes requiring cognitive control. Studies in animal models and neuroimaging in humans suggest that these effects may be related to specific actions of modafinil in the frontal cortex. The remediation of cognitive dysfunction and related neural activity may in turn form the basis of the clinical efficacy of this agent, across a range of neuropsychiatric disorders. Further investigation is necessary to confirm these initial findings, to identify specificity of these effects in the domains of neurochemistry, neuroanatomy, and cognition, and to evaluate other factors relevant to clinical use, such as the relationship of single-dose to sustained dosing regimens, and the relationship of pro-cognitive effects to clinical outcome.
References
Aguirre JA, Cintra A, Hillion J, Narvaez JA, Jansson A, Antonelli T et al (1999). A stereological study on the neuroprotective actions of acute modafinil treatment on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced nigral lesions of the male black mouse. Neurosci Lett 275: 215–218.
Akaoka H, Roussel B, Lin JS, Chouvet G, Jouvet M (1991). Effect of modafinil and amphetamine on the rat catecholaminergic neuron activity. Neurosci Lett 123: 20–22.
Antonelli T, Ferraro L, Hillion J, Tomasini MC, Rambert FA, Fuxe K (1998). Modafinil prevents glutamate cytotoxicity in cultured cortical neurons. Neuroreport 9: 4209–4213.
Aoki C, Go CG, Venkatesan C, Kurose H (1994). Perikaryal and synaptic localization of alpha 2A-adrenergic receptor-like immunoreactivity. Brain Res 650: 181–204.
Aoki C, Venkatesan C, Go CG, Forman R, Kurose H (1998). Cellular and subcellular sites for noradrenergic action in the monkey dorsolateral prefrontal cortex as revealed by the immunocytochemical localization of noradrenergic receptors and axons. Cereb Cortex 8: 269–277.
Arnsten AF (2004). Adrenergic targets for the treatment of cognitive deficits in schizophrenia. Psychopharmacology 174: 25–31.
Arnsten AF, Cai JX (1993). Postsynaptic alpha-2 receptor stimulation improves memory in aged monkeys: indirect effects of yohimbine versus direct effects of clonidine. Neurobiol Aging 14: 597–603.
Arnsten AF, Contant TA (1992). Alpha-2 adrenergic agonists decrease distractibility in aged monkeys performing the delayed response task. Psychopharmacology 108: 159–169.
Arnsten AF, Goldman-Rakic PS (1985). Alpha 2-adrenergic mechanisms in prefrontal cortex associated with cognitive decline in aged nonhuman primates. Science (New York, NY) 230: 1273–1276.
Aston-Jones G, Bloom FE (1981). Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci 1: 876–886.
Aston-Jones G, Cohen JD (2005). An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Ann Rev Neurosci 28: 403–450.
Aston-Jones G, Rajkowski J, Ivanova S, Usher M, Cohen J (1998). Neuromodulation and cognitive performance: recent studies of noradrenergic locus ceruleus neurons in behaving monkeys. Adv Pharmacol (San Diego, CA) 42: 755–759.
Aston-Jones G, Rajkowski J, Kubiak P (1997). Conditioned responses of monkey locus coeruleus neurons anticipate acquisition of discriminative behavior in a vigilance task. Neuroscience 80: 697–715.
Aston-Jones G, Rajkowski J, Kubiak P, Alexinsky T (1994). Locus coeruleus neurons in monkey are selectively activated by attended cues in a vigilance task. J Neurosci 14: 4467–4480.
Aston-Jones G, Rajkowski J, Kubiak P, Valentino RJ, Shipley MT (1996). Role of the locus coeruleus in emotional activation. Prog Brain Res 107: 379–402.
Ballon JS, Feifel D (2006). A systematic review of modafinil: potential clinical uses and mechanisms of action. J Clin Psychiatry 67: 554–566.
Baranski JV, Pigeau R, Dinich P, Jacobs I (2004). Effects of modafinil on cognitive and meta-cognitive performance. Hum Psychopharmacol 19: 323–332.
Beatty J (1982a). Phasic not tonic pupillary responses vary with auditory vigilance performance. Psychophysiology 19: 167–172.
Beatty J (1982b). Task-evoked pupillary responses, processing load, and the structure of processing resources. Psychol Bull 91: 276–292.
Beracochea D, Cagnard B, Celerier A, le Merrer J, Peres M, Pierard C (2001). First evidence of a delay-dependent working memory-enhancing effect of modafinil in mice. Neuroreport 12: 375–378.
Beracochea D, Celerier A, Borde N, Valleau M, Peres M, Pierard C (2002). Improvement of learning processes following chronic systemic administration of modafinil in mice. Pharmacol Biochem Behav 73: 723–728.
Beracochea D, Celerier A, Peres M, Pierard C (2003). Enhancement of learning processes following an acute modafinil injection in mice. Pharmacol Biochem Behav 76: 473–479.
Besset A, Chetrit M, Carlander B, Billiard M (1996). Use of modafinil in the treatment of narcolepsy: a long term follow-up study. Neurophysiol Clin 26: 60–66.
Biederman J, Swanson JM, Wigal SB, Boellner SW, Earl CQ, Lopez FA (2006). A comparison of once-daily and divided doses of modafinil in children with attention-deficit/hyperactivity disorder: a randomized, double-blind, and placebo-controlled study. J Clin Psychiatry 67: 727–735.
Biederman J, Swanson JM, Wigal SB, Kratochvil CJ, Boellner SW, Earl CQ et al (2005). Efficacy and safety of modafinil film-coated tablets in children and adolescents with attention-deficit/hyperactivity disorder: results of a randomized, double-blind, placebo-controlled, flexible-dose study. Pediatrics 116: e777–e784.
Billiard M, Besset A, Montplaisir J, Laffont F, Goldenberg F, Weill JS et al (1994). Modafinil: a double-blind multicentric study. Sleep 17: S107–S112.
Blandina P, Efoudebe M, Cenni G, Mannaioni P, Passani MB (2004). Acetylcholine, histamine, and cognition: two sides of the same coin. Learn Mem 11: 1–8.
Braver TS, Barch DM, Cohen JD (1999). Cognition and control in schizophrenia: a computational model of dopamine and prefrontal function. Biol Psychiatry 46: 312–328.
Broughton RJ, Fleming JA, George CF, Hill JD, Kryger MH, Moldofsky H et al (1997). Randomized, double-blind, placebo-controlled crossover trial of modafinil in the treatment of excessive daytime sleepiness in narcolepsy. Neurology 49: 444–451.
Bunney BS, Aghajanian GK (1976). Dopamine and norepinephrine innervated cells in the rat prefrontal cortex: pharmacological differentiation using microiontophoretic techniques. Life Sci 19: 1783–1789.
Cai JX, Ma YY, Xu L, Hu XT (1993). Reserpine impairs spatial working memory performance in monkeys: reversal by the alpha 2-adrenergic agonist clonidine. Brain Res 614: 191–196.
Carboni E, Tanda GL, Frau R, Di Chiara G (1990). Blockade of the noradrenaline carrier increases extracellular dopamine concentrations in the prefrontal cortex: evidence that dopamine is taken up in vivo by noradrenergic terminals. J Neurochem 55: 1067–1070.
Chapotot F, Pigeau R, Canini F, Bourdon L, Buguet A (2003). Distinctive effects of modafinil and d-amphetamine on the homeostatic and circadian modulation of the human waking EEG. Psychopharmacology 166: 127–138.
Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C et al (1999). Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98: 437–451.
Ciliax BJ, Drash GW, Staley JK, Haber S, Mobley CJ, Miller GW et al (1999). Immunocytochemical localization of the dopamine transporter in human brain. J Comp Neurol 409: 38–56.
Cornil CA, Balthazart J, Motte P, Massotte L, Seutin V (2002). Dopamine activates noradrenergic receptors in the preoptic area. J Neurosci 22: 9320–9330.
Crochet S, Sakai K (2003). Dopaminergic modulation of behavioral states in mesopontine tegmentum: a reverse microdialysis study in freely moving cats. Sleep 26: 801–806.
Czeisler CA, Walsh JK, Roth T, Hughes RJ, Wright KP, Kingsbury L et al (2005). Modafinil for excessive sleepiness associated with shift-work sleep disorder. N Eng J Med 353: 476–486.
Dackis CA, Kampman KM, Lynch KG, Pettinati HM, O’Brien CP (2005). A double-blind, placebo-controlled trial of modafinil for cocaine dependence. Neuropsychopharmacology 30: 205–211.
Dagan Y, Doljansky JT (2006). Cognitive performance during sustained wakefulness: a low dose of caffeine is equally effective as modafinil in alleviating the nocturnal decline. Chronobiol Int 23: 973–983.
Dauvilliers Y, Neidhart E, Billiard M, Tafti M (2002). Sexual dimorphism of the catechol-O-methyltransferase gene in narcolepsy is associated with response to modafinil. Pharmacogenomics J 2: 65–68.
de Saint Hilaire Z, Orosco M, Rouch C, Blanc G, Nicolaidis S (2001). Variations in extracellular monoamines in the prefrontal cortex and medial hypothalamus after modafinil administration: a microdialysis study in rats. Neuroreport 12: 3533–3537.
De Sereville JE, Boer C, Rambert FA, Duteil J (1994). Lack of pre-synaptic dopaminergic involvement in modafinil activity in anaesthetized mice: in vivo voltammetry studies. Neuropharmacology 33: 755–761.
DeBattista C, Lembke A, Solvason HB, Ghebremichael R, Poirier J (2004). A prospective trial of modafinil as an adjunctive treatment of major depression. J Clin Psychopharmacol 24: 87–90.
Della Marca G, Restuccia D, Rubino M, Maiese T, Tonali P (2004). Influence of modafinil on somatosensory input processing in the human brain-stem. Clin Neurophysiol 115: 919–926.
Dequardo JR (2002). Modafinil-associated clozapine toxicity. Am J Psychiatry 159: 1243–1244.
Devoto P, Flore G, Pani L, Gessa GL (2001). Evidence for co-release of noradrenaline and dopamine from noradrenergic neurons in the cerebral cortex. Mol Psychiatry 6: 657–664.
Devoto P, Flore G, Pira L, Longu G, Gessa GL (2004a). Alpha2-adrenoceptor mediated co-release of dopamine and noradrenaline from noradrenergic neurons in the cerebral cortex. J Neurochem 88: 1003–1009.
Devoto P, Flore G, Pira L, Longu G, Gessa GL (2004b). Mirtazapine-induced corelease of dopamine and noradrenaline from noradrenergic neurons in the medial prefrontal and occipital cortex. Eur J Pharmacol 487: 105–111.
Devoto P, Flore G, Saba P, Fa M, Gessa GL (2005). Stimulation of the locus coeruleus elicits noradrenaline and dopamine release in the medial prefrontal and parietal cortex. J Neurochem 92: 368–374.
Devoto P, Flore G, Vacca G, Pira L, Arca A, Casu MA et al (2003). Co-release of noradrenaline and dopamine from noradrenergic neurons in the cerebral cortex induced by clozapine, the prototype atypical antipsychotic. Psychopharmacology 167: 79–84.
Dias R, Robbins TW, Roberts AC (1996). Dissociation in prefrontal cortex of affective and attentional shifts. Nature 380: 69–72.
Dinges DF, Arora S, Darwish M, Niebler GE (2006). Pharmacodynamic effects on alertness of single doses of armodafinil in healthy subjects during a nocturnal period of acute sleep loss. Curr Med Res Opin 22: 159–167.
Durstewitz D, Seamans JK (2002). The computational role of dopamine D1 receptors in working memory. Neural Netw 15: 561–572.
Duteil J, Rambert FA, Pessonnier J, Gombert R, Assous E (1979). A possibe alpha-adrenergic mechanism for drug (CRL 40028)-induced hyperactivity. Eur J Pharmacol 59: 121–123.
Duteil J, Rambert FA, Pessonnier J, Hermant JF, Gombert R, Assous E (1990). Central alpha 1-adrenergic stimulation in relation to the behaviour stimulating effect of modafinil; studies with experimental animals. Eur J Pharmacol 180: 49–58.
Eagle DM, Tufft MR, Goodchild HL, Robbins TW (2007). Differential effects of modafinil and methylphenidate on stop-signal reaction time task performance in the rat, and interactions with the dopamine receptor antagonist cis-flupenthixol. Psychopharmacology 192: 193–206.
Edgar DM, Seidel WF (1997). Modafinil induces wakefulness without intensifying motor activity or subsequent rebound hypersomnolence in the rat. J Pharmacol Exp Ther 283: 757–769.
Ellis CM, Monk C, Simmons A, Lemmens G, Williams SC, Brammer M et al (1999). Functional magnetic resonance imaging neuroactivation studies in normal subjects and subjects with the narcoleptic syndrome. Actions of modafinil. J Sleep Res 8: 85–93.
Engber TM, Dennis SA, Jones BE, Miller MS, Contreras PC (1998a). Brain regional substrates for the actions of the novel wake-promoting agent modafinil in the rat: comparison with amphetamine. Neuroscience 87: 905–911.
Engber TM, Koury EJ, Dennis SA, Miller MS, Contreras PC, Bhat RV (1998b). Differential patterns of regional c-Fos induction in the rat brain by amphetamine and the novel wakefulness-promoting agent modafinil. Neurosci Lett 241: 95–98.
Farrow TF, Hunter MD, Haque R, Spence SA (2006). Modafinil and unconstrained motor activity in schizophrenia: double-blind crossover placebo-controlled trial. Br J Psychiatry 189: 461–462.
Fava M, Thase ME, DeBattista C (2005). A multicenter, placebo-controlled study of modafinil augmentation in partial responders to selective serotonin reuptake inhibitors with persistent fatigue and sleepiness. J Clin Psychiatry 66: 85–93.
Ferraro L, Antonelli T, O’Connor WT, Tanganelli S, Rambert F, Fuxe K (1997a). The antinarcoleptic drug modafinil increases glutamate release in thalamic areas and hippocampus. Neuroreport 8: 2883–2887.
Ferraro L, Antonelli T, O’Connor WT, Tanganelli S, Rambert FA, Fuxe K (1997b). Modafinil: an antinarcoleptic drug with a different neurochemical profile to d-amphetamine and dopamine uptake blockers. Biol Psychiatry 42: 1181–1183.
Ferraro L, Antonelli T, O’Connor WT, Tanganelli S, Rambert FA, Fuxe K (1998). The effects of modafinil on striatal, pallidal and nigral GABA and glutamate release in the conscious rat: evidence for a preferential inhibition of striato-pallidal GABA transmission. Neurosci Lett 253: 135–138.
Ferraro L, Antonelli T, Tanganelli S, O’Connor WT, Perez de la Mora M, Mendez-Franco J et al (1999). The vigilance promoting drug modafinil increases extracellular glutamate levels in the medial preoptic area and the posterior hypothalamus of the conscious rat: prevention by local GABAA receptor blockade. Neuropsychopharmacology 20: 346–356.
Ferraro L, Fuxe K, Agnati L, Tanganelli S, Tomasini MC, Antonelli T (2005). Modafinil enhances the increase of extracellular serotonin levels induced by the antidepressant drugs fluoxetine and imipramine: a dual probe microdialysis study in awake rat. Synapse (New York, NY) 55: 230–241.
Ferraro L, Fuxe K, Tanganelli S, Fernandez M, Rambert FA, Antonelli T (2000). Amplification of cortical serotonin release: a further neurochemical action of the vigilance-promoting drug modafinil. Neuropharmacology 39: 1974–1983.
Ferraro L, Fuxe K, Tanganelli S, Tomasini MC, Rambert FA, Antonelli T (2002). Differential enhancement of dialysate serotonin levels in distinct brain regions of the awake rat by modafinil: possible relevance for wakefulness and depression. J Neurosci Res 68: 107–112.
Ferraro L, Tanganelli S, Fuxe K, Bebe BW, Tomasini MC, Rambert FA et al (2001). Modafinil does not affect serotonin efflux from rat frontal cortex synaptosomes: comparison with known serotonergic drugs. Brain Res 894: 307–310.
Ferraro L, Tanganelli S, O’Connor WT, Antonelli T, Rambert F, Fuxe K (1996). The vigilance promoting drug modafinil decreases GABA release in the medial preoptic area and in the posterior hypothalamus of the awake rat: possible involvement of the serotonergic 5-HT3 receptor. Neurosci Lett 220: 5–8.
Florence G, Bonnier R, Plagnes D, Pierard C, Satabin P, Peres M et al (2000). Effect of modafinil on cerebral blood flow of anaesthetised rats. Comparison with amphetamine. Exp Brain Res 135: 552–556.
Fuxe K, Janson AM, Rosen L, Finnman UB, Tanganelli S, Morari M et al (1992). Evidence for a protective action of the vigilance promoting drug modafinil on the MPTP-induced degeneration of the nigrostriatal dopamine neurons in the black mouse: an immunocytochemical and biochemical analysis. Exp Brain Res 88: 117–130.
Gallopin T, Luppi PH, Rambert FA, Frydman A, Fort P (2004). Effect of the wake-promoting agent modafinil on sleep-promoting neurons from the ventrolateral preoptic nucleus: an in vitro pharmacologic study. Sleep 27: 19–25.
Gao WJ, Krimer LS, Goldman-Rakic PS (2001). Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl AcadSci USA 98: 295–300.
Geracitano R, Federici M, Prisco S, Bernardi G, Mercuri NB (2004). Inhibitory effects of trace amines on rat midbrain dopaminergic neurons. Neuropharmacology 46: 807–814.
Gill M, Haerich P, Westcott K, Godenick KL, Tucker JA (2006). Cognitive performance following modafinil versus placebo in sleep-deprived emergency physicians: a double-blind randomized crossover study. Acad Emerg Med 13: 158–165.
Gold LH, Balster RL (1996). Evaluation of the cocaine-like discriminative stimulus effects and reinforcing effects of modafinil. Psychopharmacology 126: 286–292.
Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV (2004). Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology 174: 3–16.
Greenhill LL, Biederman J, Boellner SW, Rugino TA, Sangal RB, Earl CQ et al (2006). A randomized, double-blind, placebo-controlled study of modafinil film-coated tablets in children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 45: 503–511.
Haas H, Panula P (2003). The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev 4: 121–130.
Harsh JR, Hayduk R, Rosenberg R, Wesnes KA, Walsh JK, Arora S et al (2006). The efficacy and safety of armodafinil as treatment for adults with excessive sleepiness associated with narcolepsy. Curr Med Res Opin 22: 761–774.
Hart CL, Haney M, Vosburg SK, Comer SD, Gunderson E, Foltin RW (2006). Modafinil attenuates disruptions in cognitive performance during simulated night-shift work. Neuropsychopharmacology 31: 1526–1536.
Hellriegel ET, Arora S, Nelson M, Robertson Jr P (2001). Steady-state pharmacokinetics and tolerability of modafinil given alone or in combination with methylphenidate in healthy volunteers. J Clin Pharmacol 41: 895–904.
Hellriegel ET, Arora S, Nelson M, Robertson Jr P (2002). Steady-state pharmacokinetics and tolerability of modafinil administered alone or in combination with dextroamphetamine in healthy volunteers. J Clin Pharmacol 42: 450–460.
Hermant JF, Rambert FA, Duteil J (1991). Awakening properties of modafinil: effect on nocturnal activity in monkeys (Macaca mulatta) after acute and repeated administration. Psychopharmacology 103: 28–32.
Hieble JP, Bondinell WE, Ruffolo Jr RR (1995). Alpha- and beta-adrenoceptors: from the gene to the clinic. 1. Molecular biology and adrenoceptor subclassification. J Med Chem 38: 3415–3444.
Hirshkowitz M, Black JE, Wesnes K, Niebler G, Arora S, Roth T (2006). Adjunct armodafinil improves wakefulness and memory in obstructive sleep apnea/hypopnea syndrome. Respir Med 101: 616–627.
Hou RH, Freeman C, Langley RW, Szabadi E, Bradshaw CM (2005). Does modafinil activate the locus coeruleus in man? Comparison of modafinil and clonidine on arousal and autonomic functions in human volunteers. Psychopharmacology 181: 537–549.
Hunter MD, Ganesan V, Wilkinson ID, Spence SA (2006). Impact of modafinil on prefrontal executive function in schizophrenia. Am J Psychiatry 163: 2184–2186.
Ishizuka T, Sakamoto Y, Sakurai T, Yamatodani A (2003). Modafinil increases histamine release in the anterior hypothalamus of rats. Neurosci Lett 339: 143–146.
Jasinski DR (2000). An evaluation of the abuse potential of modafinil using methylphenidate as a reference. J Psychopharmacol 14: 53–60.
Jones SR, Gainetdinov RR, Hu XT, Cooper DC, Wightman RM, White FJ et al (1999). Loss of autoreceptor functions in mice lacking the dopamine transporter. Nat Neurosci 2: 649–655.
Kawahara H, Kawahara Y, Westerink BH (2001). The noradrenaline-dopamine interaction in the rat medial prefrontal cortex studied by multi-probe microdialysis. Eur J Pharmacol 418: 177–186.
Korotkova TM, Klyuch BP, Ponomarenko AA, Lin JS, Haas HL, Sergeeva OA (2006). Modafinil inhibits rat midbrain dopaminergic neurons through D2-like receptors. Neuropharmacology 52: 626–633.
Krazem A, Beracochea D, Jaffard R (1995). Effects of mammillary bodies and mediodorsal thalamic lesions on the acquisition and retention of a learning set in mice: paradoxical effect of the intersession interval. Behav Brain Res 67: 51–58.
Lewis DA, Morrison JH (1989). Noradrenergic innervation of monkey prefrontal cortex: a dopamine-beta-hydroxylase immunohistochemical study. J Comp Neurol 282: 317–330.
Li BM, Mao ZM, Wang M, Mei ZT (1999). Alpha-2 adrenergic modulation of prefrontal cortical neuronal activity related to spatial working memory in monkeys. Neuropsychopharmacology 21: 601–610.
Lin JS, Gervasoni D, Hou Y, Vanni-Mercier G, Rambert F, Frydman A et al (2000). Effects of amphetamine and modafinil on the sleep/wake cycle during experimental hypersomnia induced by sleep deprivation in the cat. J Sleep Res 9: 89–96.
Lin JS, Hou Y, Jouvet M (1996). Potential brain neuronal targets for amphetamine-, methylphenidate-, and modafinil-induced wakefulness, evidenced by c-fos immunocytochemistry in the cat. Proc Natl Acad Sci USA 93: 14128–14133.
Lin JS, Roussel B, Akaoka H, Fort P, Debilly G, Jouvet M (1992). Role of catecholamines in the modafinil and amphetamine induced wakefulness, a comparative pharmacological study in the cat. Brain Res 591: 319–326.
MacDonald JR, Hill JD, Tarnopolsky MA (2002). Modafinil reduces excessive somnolence and enhances mood in patients with myotonic dystrophy. Neurology 59: 1876–1880.
Madras BK, Xie Z, Lin Z, Jassen A, Panas H, Lynch L et al (2006). Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J Pharmacol Exp Ther 319: 561–569.
Malcolm R, Swayngim K, Donovan JL, DeVane CL, Elkashef A, Chiang N et al (2006). Modafinil and cocaine interactions. Am J Drug Alcohol Abuse 32: 577–587.
Malenka RC, Nicoll RA (1986). Dopamine decreases the calcium-activated afterhyperpolarization in hippocampal CA1 pyramidal cells. Brain Res 379: 210–215.
Marek GJ, Aghajanian GK (1999). 5-HT2A receptor or alpha1-adrenoceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur J Pharmacol 367: 197–206.
Meunier M, Jaffard R, Destrade C (1991). Differential involvement of anterior and posterior cingulate cortices in spatial discriminative learning in a T-maze in mice. Behav Brain Res 44: 133–143.
Mignot E, Nishino S, Guilleminault C, Dement WC (1994). Modafinil binds to the dopamine uptake carrier site with low affinity. Sleep 17: 436–437.
Mignot E, Renaud A, Nishino S, Arrigoni J, Guilleminault C, Dement WC (1993). Canine cataplexy is preferentially controlled by adrenergic mechanisms: evidence using monoamine selective uptake inhibitors and release enhancers. Psychopharmacology 113: 76–82.
Millan MJ, Lejeune F, Gobert A (2000). Reciprocal autoreceptor and heteroreceptor control of serotonergic, dopaminergic and noradrenergic transmission in the frontal cortex: relevance to the actions of antidepressant agents. J Psychopharmacol 14: 114–138.
Miller GM, Verrico CD, Jassen A, Konar M, Yang H, Panas H et al (2005). Primate trace amine receptor 1 modulation by the dopamine transporter. J Pharmacol Exp Ther 313: 983–994.
Mitler MM, Harsh J, Hirshkowitz M, Guilleminault C (2000). Long-term efficacy and safety of modafinil (PROVIGIL(R)) for the treatment of excessive daytime sleepiness associated with narcolepsy. Sleep Med 1: 231–243.
Morgan RE, Crowley JM, Smith RH, Laroche RB, Dopheide MM (2007). Modafinil improves attention, inhibitory control, and reaction time in healthy, middle-aged rats. Pharmacol Biochem Behav 86: 531–541.
Moron JA, Brockington A, Wise RA, Rocha BA, Hope BT (2002). Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci 22: 389–395.
Muller U, Steffenhagen N, Regenthal R, Bublak P (2004). Effects of modafinil on working memory processes in humans. Psychopharmacology 177: 161–169.
Muly III EC, Szigeti K, Goldman-Rakic PS (1998). D1 receptor in interneurons of macaque prefrontal cortex: distribution and subcellular localization. J Neurosci 18: 10553–10565.
Murillo-Rodriguez E, Haro R, Palomero-Rivero M, Millan-Aldaco D, Drucker-Colin R (2007). Modafinil enhances extracellular levels of dopamine in the nucleus accumbens and increases wakefulness in rats. Behav Brain Res 176: 353–357.
Myrick H, Malcolm R, Taylor B, LaRowe S (2004). Modafinil: preclinical, clinical, and post-marketing surveillance—a review of abuse liability issues. Ann Clin Psychiatry 16: 101–109.
Nagels G, D’Hooghe MB, Vleugels L, Kos D, Despontin M, De Deyn PP (2007). P300 and treatment effect of modafinil on fatigue in multiple sclerosis. J Clin Neurosci 14: 33–40.
Narendran R, Young CM, Valenti AM, Nickolova MK, Pristach CA (2002). Is psychosis exacerbated by modafinil? Arch Gen Psychiatry 59: 292–293.
Nathaniel-James DA, Fletcher P, Frith CD (1997). The functional anatomy of verbal initiation and suppression using the Hayling Test. Neuropsychologia 35: 559–566.
Nathaniel-James DA, Frith CD (2002). The role of the dorsolateral prefrontal cortex: evidence from the effects of contextual constraint in a sentence completion task. NeuroImage 16: 1094–1102.
Nishino S (2003). The hypocretin/orexin system in health and disease. Biol Psychiatry 54: 87–95.
Nishino S, Fruhstorfer B, Arrigoni J, Guilleminault C, Dement WC, Mignot E (1993). Further characterization of the alpha-1 receptor subtype involved in the control of cataplexy in canine narcolepsy. J Pharmacol Exp Ther 264: 1079–1084.
Oskooilar N (2005). A case of premature ventricular contractions with modafinil. Am J Psychiatry 162: 1983–1984.
Passani MB, Bacciottini L, Mannaioni PF, Blandina P (2000). Central histaminergic system and cognition. Neurosci Biobehav Rev 24: 107–113.
Perez de la Mora M, Aguilar-Garcia A, Ramon-Frias T, Ramirez-Ramirez R, Mendez-Franco J, Rambert F et al (1999). Effects of the vigilance promoting drug modafinil on the synthesis of GABA and glutamate in slices of rat hypothalamus. Neurosci Lett 259: 181–185.
Pierard C, Liscia P, Valleau M, Drouet I, Chauveau F, Huart B et al (2006). Modafinil-induced modulation of working memory and plasma corticosterone in chronically-stressed mice. Pharmacol Biochem Behav 83: 1–8.
Pierard C, Satabin P, Lagarde D, Barrere B, Guezennec CY, Menu JP et al (1995). Effects of a vigilance-enhancing drug, modafinil, on rat brain metabolism: a 2D COSY 1H-NMR study. Brain Res 693: 251–256.
Pierre JM, Peloian JH, Wirshing DA, Wirshing WC, Marder SR (2005). A double-blind placebo-controlled trial of modafinil for negative symptoms in schizophrenia. Neuropsychopharmacology 30(Suppl 1): S207.
Pigeau R, Naitoh P, Buguet A, McCann C, Baranski J, Taylor M et al (1995). Modafinil, d-amphetamine and placebo during 64 h of sustained mental work. I. Effects on mood, fatigue, cognitive performance and body temperature. J Sleep Res 4: 212–228.
Rambert FA, Pessionier J, Duteil J (1993). Modafinil-, amphetamine- and methylphenidate-induced hyperactivities in mice involve different mechanisms. Eur J Pharmacol 183: 455–456.
Randall DC, Fleck NL, Shneerson JM, File SE (2004). The cognitive-enhancing properties of modafinil are limited in non-sleep-deprived middle-aged volunteers. Pharmacol Biochem Behav 77: 547–555.
Randall DC, Shneerson JM, File SE (2005a). Cognitive effects of modafinil in student volunteers may depend on IQ. Pharmacol Biochem Behav 82: 133–139.
Randall DC, Shneerson JM, Plaha KK, File SE (2003). Modafinil affects mood, but not cognitive function, in healthy young volunteers. Hum Psychopharmacol 18: 163–173.
Randall DC, Viswanath A, Bharania P, Elsabagh SM, Hartley DE, Shneerson JM et al (2005b). Does modafinil enhance cognitive performance in young volunteers who are not sleep-deprived? J Clin Psychopharmacol 25: 175–179.
Ranjan S, Chandra PS (2005). Modafinil-induced irritability and aggression? A report of 2 bipolar patients. J Clin Psychopharmacol 25: 628–629.
Richer F, Beatty J (1987). Contrasting effects of response uncertainty on the task-evoked pupillary response and reaction time. Psychophysiology 24: 258–262.
Robertson P, DeCory HH, Madan A, Parkinson A (2000). In vitro inhibition and induction of human hepatic cytochrome P450 enzymes by modafinil. Drug Metab Dispos 28: 664–671.
Robertson Jr P, Hellriegel ET (2003). Clinical pharmacokinetic profile of modafinil. Clin Pharmacokinet 42: 123–137.
Robertson Jr P, Hellriegel ET, Arora S, Nelson M (2002a). Effect of modafinil at steady state on the single-dose pharmacokinetic profile of warfarin in healthy volunteers. J Clin Pharmacol 42: 205–214.
Robertson Jr P, Hellriegel ET, Arora S, Nelson M (2002b). Effect of modafinil on the pharmacokinetics of ethinyl estradiol and triazolam in healthy volunteers. Clin Pharmacol Ther 71: 46–56.
Rosenthal MH, Bryant SL (2004). Benefits of adjunct modafinil in an open-label, pilot study in patients with schizophrenia. Clin Neuropharmacol 27: 38–43.
Roth T, White D, Schmidt-Nowara W, Wesnes KA, Niebler G, Arora S et al (2006). Effects of armodafinil in the treatment of residual excessive sleepiness associated with obstructive sleep apnea/hypopnea syndrome: a 12-week, multicenter, double-blind, randomized, placebo-controlled study in nCPAP-adherent adults. Clin Ther 28: 689–706.
Rugino TA, Copley TC (2001). Effects of modafinil in children with attention-deficit/hyperactivity disorder: an open-label study. J Am Acad Child Adolesc Psychiatry 40: 230–235.
Rugino TA, Samsock TC (2003). Modafinil in children with attention-deficit hyperactivity disorder. Pediatr Neurol 29: 136–142.
Saletu M, Anderer P, Saletu-Zyhlarz GM, Mandl M, Arnold O, Zeitlhofer J et al (2004). EEG-tomographic studies with LORETA on vigilance differences between narcolepsy patients and controls and subsequent double-blind, placebo-controlled studies with modafinil. J Neurol 251: 1354–1363.
Saletu M, Anderer P, Semlitsch HV, Saletu-Zyhlarz GM, Mandl M, Zeitlhofer J et al (2007). Low-resolution brain electromagnetic tomography (LORETA) identifies brain regions linked to psychometric performance under modafinil in narcolepsy. Psychiatry Res 154: 69–84.
Samuels ER, Hou RH, Langley RW, Szabadi E, Bradshaw CM (2006). Comparison of pramipexole and modafinil on arousal, autonomic, and endocrine functions in healthy volunteers. J Psychopharmacol 20: 756–770.
Sangal RB, Sangal JM, Belisle C (1999a). Longer auditory and visual P300 latencies in patients with narcolepsy. Clin Electroencephalogr 30: 28–32.
Sangal RB, Sangal JM, Belisle C (1999b). Visual P300 latency predicts treatment response to modafinil in patients with narcolepsy. Clin Neurophysiol 110: 1041–1047.
Sawaguchi T (1998). Attenuation of delay-period activity of monkey prefrontal neurons by an alpha2-adrenergic antagonist during an oculomotor delayed-response task. J Neurophysiol 80: 2200–2205.
Sawaguchi T, Goldman-Rakic PS (1991). D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science 251: 947–950.
Sawaguchi T, Goldman-Rakic PS (1994). The role of D1-dopamine receptor in working memory: local injections of dopamine antagonists into the prefrontal cortex of rhesus monkeys performing an oculomotor delayed-response task. J Neurophysiol 71: 515–528.
Scammell TE, Estabrooke IV, McCarthy MT, Chemelli RM, Yanagisawa M, Miller MS et al (2000). Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci 20: 8620–8628.
Schwartz JR, Hirshkowitz M, Erman MK, Schmidt-Nowara W (2003). Modafinil as adjunct therapy for daytime sleepiness in obstructive sleep apnea: a 12-week, open-label study. Chest 124: 2192–2199.
Schwartz JR, Nelson MT, Schwartz ER, Hughes RJ (2004). Effects of modafinil on wakefulness and executive function in patients with narcolepsy experiencing late-day sleepiness. Clin Neuropharmacol 27: 74–79.
Schwertner HA, Kong SB (2005). Determination of modafinil in plasma and urine by reversed phase high-performance liquid-chromatography. J Pharm Biomed Anal 37: 475–479.
Seamans JK, Yang CR (2004). The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol 74: 1–58.
Sesack SR, Hawrylak VA, Guido MA, Levey AI (1998). Cellular and subcellular localization of the dopamine transporter in rat cortex. Adv Pharmacol 42: 171–174.
Sevy S, Rosenthal MH, Alvir J, Meyer S, Visweswaraiah H, Gunduz-Bruce H et al (2005). Double-blind, placebo-controlled study of modafinil for fatigue and cognition in schizophrenia patients treated with psychotropic medications. J Clin Psychiatry 66: 839–843.
Shelton J, Nishino S, Vaught J, Dement WC, Mignot E (1995). Comparative effects of modafinil and amphetamine on daytime sleepiness and cataplexy of narcoleptic dogs. Sleep 18: 817–826.
Simon P, Hemet C, Ramassamy C, Costentin J (1995). Non-amphetaminic mechanism of stimulant locomotor effect of modafinil in mice. Eur Neuropsychopharmacol 5: 509–514.
Simon P, Panissaud C, Costentin J (1994). The stimulant effect of modafinil on wakefulness is not associated with an increase in anxiety in mice. A comparison with dexamphetamine. Psychopharmacology 114: 597–600.
Smiley JF, Goldman-Rakic PS (1993). Heterogeneous targets of dopamine synapses in monkey prefrontal cortex demonstrated by serial section electron microscopy: a laminar analysis using the silver-enhanced diaminobenzidine sulfide (SEDS) immunolabeling technique. Cereb Cortex 3: 223–238.
Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS (1994). D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. 91: 5720–5724.
Souliere F, Urbain N, Gervasoni D, Schmitt P, Guillemort C, Fort P et al (2000). Single-unit and polygraphic recordings associated with systemic or local pharmacology: a multi-purpose stereotaxic approach for the awake, anaesthetic-free, and head-restrained rat. J Neurosci Res 61: 88–100.
Spence SA, Green RD, Wilkinson ID, Hunter MD (2005). Modafinil modulates anterior cingulate function in chronic schizophrenia. Br J Psychiatry 187: 55–61.
Stone EA, Cotecchia S, Lin Y, Quartermain D (2002a). Role of brain alpha 1B-adrenoceptors in modafinil-induced behavioral activity. Synapse 46: 269–270.
Stone EA, Lin Y, Suckow RF, Quartermain D (2002b). Stress-induced subsensitivity to modafinil and its prevention by corticosteroids. Pharmacol Biochem Behav 73: 971–978.
Swanson JM, Greenhill LL, Lopez FA, Sedillo A, Earl CQ, Jiang JG et al (2006). Modafinil film-coated tablets in children and adolescents with attention-deficit/hyperactivity disorder: results of a randomized, double-blind, placebo-controlled, fixed-dose study followed by abrupt discontinuation. J Clin Psychiatry 67: 137–147.
Taneja I, Haman K, Shelton RC, Robertson D (2007). A randomized, double-blind, crossover trial of modafinil on mood. J Clin Psychopharmacol 27: 76–79.
Tanganelli S, Ferraro L, Bianchi C, Fuxe K (1994). 6-hydroxy-dopamine treatment counteracts the reduction of cortical GABA release produced by the vigilance promoting drug modafinil in the awake freely moving guinea-pig. Neurosci Lett 171: 201–204.
Tanganelli S, Fuxe K, Ferraro L, Janson AM, Bianchi C (1992). Inhibitory effects of the psychoactive drug modafinil on gamma-aminobutyric acid outflow from the cerebral cortex of the awake freely moving guinea-pig. Possible involvement of 5-hydroxytryptamine mechanisms. Naunyn-Schmiedeberg's Archives of Pharmacology 345: 461–465.
Tanganelli S, Perez de la Mora M, Ferraro L, Mendez-Franco J, Beani L, Rambert FA et al (1995). Modafinil and cortical gamma-aminobutyric acid outflow. Modulation by 5-hydroxytryptamine neurotoxins. Eur J Pharmacol 273: 63–71.
Tassin JP, Bockaert J, Blanc G, Stinus L, Thierry AM, Lavielle S et al (1978). Topographical distribution of dopaminergic innervation and dopaminergic receptors of the anterior cerebral cortex of the rat. Brain Res 154: 241–251.
Taylor FB, Russo J (2000). Efficacy of modafinil compared to dextroamphetamine for the treatment of attention deficit hyperactivity disorder in adults. J Child Adolesc Psychopharmacol 10: 311–320.
Thase ME, Fava M, DeBattista C, Arora S, Hughes RJ (2006). Modafinil augmentation of SSRI therapy in patients with major depressive disorder and excessive sleepiness and fatigue: a 12-week, open-label, extension study. CNS Spectrums 11: 93–102.
Thomas RJ, Kwong K (2006). Modafinil activates cortical and subcortical sites in the sleep-deprived state. Sleep 29: 1471–1481.
Touret M, Sallanon-Moulin M, Fages C, Roudier V, Didier-Bazes M, Roussel B et al (1994). Effects of modafinil-induced wakefulness on glutamine synthetase regulation in the rat brain. Brain Res Mol Brain Res 26: 123–128.
Touret M, Sallanon-Moulin M, Jouvet M (1995). Awakening properties of modafinil without paradoxical sleep rebound: comparative study with amphetamine in the rat. Neurosci Lett 189: 43–46.
Tseng YL, Uralets V, Lin CT, Kuo FH (2005). Detection of modafinil in human urine by gas chromatography-mass spectrometry. J Pharm Biomed Anal 39: 1042–1045.
Turner DC, Clark L, Dowson J, Robbins TW, Sahakian BJ (2004a). Modafinil improves cognition and response inhibition in adult attention-deficit/hyperactivity disorder. Biol Psychiatry 55: 1031–1040.
Turner DC, Clark L, Pomarol-Clotet E, McKenna P, Robbins TW, Sahakian BJ (2004b). Modafinil improves cognition and attentional set shifting in patients with chronic schizophrenia. Neuropsychopharmacology 29: 1363–1373.
Turner DC, Robbins TW, Clark L, Aron AR, Dowson J, Sahakian BJ (2003). Cognitive enhancing effects of modafinil in healthy volunteers. Psychopharmacology 165: 260–269.
Usher M, Cohen JD, Servan-Schreiber D, Rajkowski J, Aston-Jones G (1999). The role of locus coeruleus in the regulation of cognitive performance. Science 283: 549–554.
US Modafinil in Narcolepsy Multicenter Study Group (1998). Randomized trial of modfinil for the treatment of pathological somnolence in narcolepsy. Ann Neurol 43: 88–97.
US Modafinil in Narcolepsy Multicenter Study Group (2000). Randomized trial of modafinil as a treatment for the excessive daytime somnolence of narcolepsy. Neurology 54: 1166–1175.
Vorspan F, Warot D, Consoli A, Cohen D, Mazet P (2005). Mania in a boy treated with modafinil for narcolepsy. Am J Psychiatry 162: 813–814.
Walsh JK, Randazzo AC, Stone KL, Schweitzer PK (2004). Modafinil improves alertness, vigilance, and executive function during simulated night shifts. Sleep 27: 434–439.
Ward CP, Harsh JR, York KM, Stewart KL, McCoy JG (2004). Modafinil facilitates performance on a delayed nonmatching to position swim task in rats. Pharmacol Biochem Behav 78: 735–741.
Waters KA, Burnham KE, O’Connor D, Dawson GR, Dias R (2005). Assessment of modafinil on attentional processes in a five-choice serial reaction time test in the rat. J Psychopharmacol 19: 149–158.
Wesensten NJ (2006). Effects of modafinil on cognitive performance and alertness during sleep deprivation. Curr Pharm Des 12: 2457–2471.
Wesensten NJ, Belenky G, Kautz MA, Thorne DR, Reichardt RM, Balkin TJ (2002). Maintaining alertness and performance during sleep deprivation: modafinil versus caffeine. Psychopharmacology 159: 238–247.
Wesensten NJ, Killgore WD, Balkin TJ (2005). Performance and alertness effects of caffeine, dextroamphetamine, and modafinil during sleep deprivation. J Sleep Res 14: 255–266.
Wieland HA, Soll RM, Doods HN, Stenkamp D, Hurnaus R, Lammle B et al (2002). The SK-N-MC cell line expresses an orexin binding site different from recombinant orexin 1-type receptor. Eur J Biochemistry 269: 1128–1135.
Williams GV, Castner SA (2006). Under the curve: critical issues for elucidating D1 receptor function in working memory. Neuroscience 139: 263–276.
Williams SM, Goldman-Rakic PS (1993). Characterization of the dopaminergic innervation of the primate frontal cortex using a dopamine-specific antibody. Cereb Cortex 3: 199–222.
Willie JT, Renthal W, Chemelli RM, Miller MS, Scammell TE, Yanagisawa M et al (2005). Modafinil more effectively induces wakefulness in orexin-null mice than in wild-type littermates. Neuroscience 130: 983–995.
Wisor JP, Dement WC, Aimone L, Williams M, Bozyczko-Coyne D (2006). Armodafinil, the R-enantiomer of modafinil: wake-promoting effects and pharmacokinetic profile in the rat. Pharmacol Biochem Behav 85: 492–499.
Wisor JP, Eriksson KS (2005). Dopaminergic-adrenergic interactions in the wake promoting mechanism of modafinil. Neuroscience 132: 1027–1034.
Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM (2001). Dopaminergic role in stimulant-induced wakefulness. J Neurosci 21: 1787–1794.
Wolf J, Fiedler U, Anghelescu I, Schwertfeger N (2006). Manic switch in a patient with treatment-resistant bipolar depression treated with modafinil. J Clin Psychiatry 67: 1817.
Wong YN, King SP, Laughton WB, McCormick GC, Grebow PE (1998a). Single-dose pharmacokinetics of modafinil and methylphenidate given alone or in combination in healthy male volunteers. J Clin Pharmacol 38: 276–282.
Wong YN, King SP, Simcoe D, Gorman S, Laughton W, McCormick GC et al (1999a). Open-label, single-dose pharmacokinetic study of modafinil tablets: influence of age and gender in normal subjects. J Clin Pharmacol 39: 281–288.
Wong YN, Simcoe D, Hartman LN, Laughton WB, King SP, McCormick GC et al (1999b). A double-blind, placebo-controlled, ascending-dose evaluation of the pharmacokinetics and tolerability of modafinil tablets in healthy male volunteers. J Clin Pharmacol 39: 30–40.
Wong YN, Wang L, Hartman L, Simcoe D, Chen Y, Laughton W et al (1998b). Comparison of the single-dose pharmacokinetics and tolerability of modafinil and dextroamphetamine administered alone or in combination in healthy male volunteers. J Clin Pharmacol 38: 971–978.
Xie Z, Miller GM (2007). Trace Amine-Associated Receptor 1 is a Modulator of the Dopamine Transporter. J Pharmacol Exp Ther 321: 128–136.
Xie Z, Westmoreland S, Bahn ME, Chen GL, Yang H, Vallender E et al (2007). Rhesus monkey trace amine-associated receptor 1 signaling: enhancement by monoamine transporters and attenuation by the D2 autoreceptor in vitro. J Pharmacol Exp Ther 321: 116–127.
Zhang WP, Ouyang M, Thomas SA (2004). Potency of catecholamines and other L-tyrosine derivatives at the cloned mouse adrenergic receptors. Neuropharmacology 47: 438–449.
Zifko UA, Rupp M, Schwarz S, Zipko HT, Maida EM (2002). Modafinil in treatment of fatigue in multiple sclerosis. Results of an open-label study. J Neurol 249: 983–987.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICTS OF INTEREST
Dr Minzenberg and Dr Carter have received research funding from Cephalon, a manufacturer of modafinil and armodafinil. Dr Carter has served as a consultant for Pfizer, Hoffman La Roche, and Lilly. Dr Minzenberg holds stock in Elan Pharmaceuticals. No support was received from these companies in the background research for, or preparation of, the present manuscript. Funding to support this work was received from a Translational Clinical Scientist Award from the Burroughs Wellcome Foundation, and MH59883 and MH066629 from the NIMH, all to Dr Carter.
Rights and permissions
About this article
Cite this article
Minzenberg, M., Carter, C. Modafinil: A Review of Neurochemical Actions and Effects on Cognition. Neuropsychopharmacol 33, 1477–1502 (2008). https://doi.org/10.1038/sj.npp.1301534
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301534
Keywords
This article is cited by
-
G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders
Signal Transduction and Targeted Therapy (2023)
-
Open-label placebo treatment does not enhance cognitive abilities in healthy volunteers
Scientific Reports (2023)
-
Pharmacologic Management of Cognitive Disengagement Syndrome (CDS) and Implications for Attention-Deficit/Hyperactivity Disorder (ADHD) Treatment: Emerging Treatments and Recommendations for Future Research
CNS Drugs (2023)
-
Selenium alleviates modafinil-induced neurobehavioral toxicity in rat via PI3K/Akt/mTOR/GSK3B signaling pathway and suppression of oxidative stress and apoptosis: in vivo and in silico study
Environmental Science and Pollution Research (2023)
-
Comparison of Solriamfetol and Modafinil on Arousal and Anxiety-Related Behaviors in Narcoleptic Mice
Neurotherapeutics (2023)
