
Abstract
The limited success of genetic studies of major depression has raised questions concerning the definition of genetically relevant phenotypes. This paper presents strategies to improve the phenotypic definition of major depression by proposing endophenotypes at two levels: First, dissecting the depressive phenotype into key components results in narrow definitions of putative psychopathological endophenotypes: mood bias toward negative emotions, impaired reward function, impaired learning and memory, neurovegetative signs, impaired diurnal variation, impaired executive cognitive function, psychomotor change, and increased stress sensitivity. A review of the recent literature on neurobiological and genetic findings associated with these components is given. Second, the most consistent heritable biological markers of major depression are proposed as biological endophenotypes for genetic studies: REM sleep abnormalities, functional and structural brain abnormalities, dysfunctions in serotonergic, catecholaminergic, hypothalamic-pituitary-adrenocortical axis, and CRH systems, and intracellular signal transduction endophenotypes. The associations among the psychopathological and biological endophenotypes are discussed with respect to specificity, temporal stability, heritability, familiality, and clinical and biological plausibility. Finally, the case is made for the development of a new classification system in order to reduce the heterogeneity of depression representing a major impediment to elucidating the genetic and neurobiological basis of this common, severe, and often life-threatening illness.
Similar content being viewed by others

INTRODUCTION
Scientific advances suggest that the time is at hand to begin to elucidate the genetic basis of mood disorders: (1) detection of genes for brain diseases such as Huntington's disease and Alzheimer's disease; (2) dramatic developments in molecular genetics including the human genome project and increasing availability of genetic markers throughout the genome; and (3) consistent evidence from twin and family studies that genes are substantially involved in the susceptibility to mood disorders. However, despite costly candidate gene association studies and genome-wide linkage scans, no genes for major depression have been consistently identified. Fortunately, in certain families, there is preliminary evidence that recurrent, early-onset major depression is linked to a region containing the CREB1 gene. This gene constitutes an attractive candidate as a susceptibility allele for major depression because CREB1 plays major roles in neuronal plasticity, cognition, and memory (Zubenko et al, 2002).
The limited success of genetic studies of complex disorders has resulted in a considerable debate regarding the reasons for the failure in the past, and the best methodological approach to take in the future. Questions have been raised concerning both the definition of genetically relevant phenotypes and the number and nature of the underlying genes.
The use of the current classification schemas including DSM-IV undoubtedly contributes to the difficulties in finding genes for psychiatric disorders. They are based on clusters of symptoms and characteristics of clinical course that do not necessarily describe homogenous disorders, and rather reflect common final pathways of different pathophysiological processes (Charney et al, 2002). Moreover, it has been argued that a continuum of depressive symptoms exists from normal to pathological, and that clinical symptomatology does not point to a simple categorical classification (Angst and Dobler-Mikola, 1984; Angst and Merikangas, 2001). Finally, mood and anxiety disorders as defined by the DSM show high comorbidity and substantial symptomatic fluidity with frequent changes of diagnostic subtypes over time (Angst and Dobler-Mikola, 1985; Angst et al, 1990; 2000; Merikangas et al, 2003).
Moreover, genes and behaviors may not be associated on a simplistic, one-to-one basis; the true relationship between a gene and a behavior is probably more akin to chaos theory's ‘sensitive dependence on initial conditions’. For example, there is presumably no gene for ‘language’; there are a number of genes that pattern the embryonic brain in such a way as to facilitate, and allow the physiological processes necessary for, language acquisition. In a similar manner, no gene has been found to singularly code for a human psychiatric condition. Indeed, it is unlikely that any single gene does code for a psychiatric condition per se, but rather that susceptibility genes interacting with developmental factors, both day-to-day and profound environmental events, epigenetic DNA modifications, and possible entirely stochastic mechanisms eventually lead to the development of normal and abnormal human behaviors (Petronis, 2001; Glazier et al, 2002).
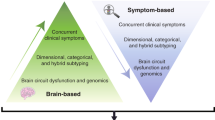
In this paper, we will present strategies to overcome the methodological difficulties mentioned above with respect to the elucidation of the genetic basis of major depressive disorder (MDD) by proposing putative endophenotypes. The term ‘endophenotype’ was described as an internal phenotype (ie not obvious to the unaided eye) that fills the gap between available descriptors and between the gene and the elusive disease process (Gottesman and Shields, 1973), and therefore may help to resolve questions about etiological models. The endophenotype concept was based on the assumption that the number of genes involved in the variations of endophenotypes representing relatively straightforward and putatively more elementary phenomena (as opposed to behavioral macros) are fewer than those involved in producing a psychiatric diagnostic entity (Gottesman and Gould, 2003). Endophenotypes provide a means for identifying the ‘downstream’ traits of clinical phenotypes, as well as the ‘upstream’ consequence of genes. The methods available to identify endophenotypes include neuropsychological, cognitive, neurophysiological, neuroanatomical, and biochemical measures (Figures 1 and 2). The evaluation of endophenotypes is based on the following criteria (Tsuang et al, 1993):

Example of how neuroanatomical abnormalities may relate to candidate genes and to key components of major depression. Some of the key components have a greater potential to serve as endophenotypes than others (see Table 1). Not all functional directions are indicated for the purpose of clarity of the figure.

Example of how neurochemical abnormalities may relate to candidate genes and to key components of major depression. Some of the key components have a greater potential to serve as endophenotypes than others (see Table 1). Not all functional directions are indicated for the purpose of clarity of the figure.
Specificity: The endophenotype is more strongly associated with the disease of interest than with other psychiatric conditions.
State-independence: The endophenotype is stable over time and not an epiphenomenon of the illness or its treatment.
Heritability: Variance in the endophenotype is associated with genetic variance.
Familial association: The endophenotype is more prevalent among the relatives of ill probands compared with an appropriate control group.
Cosegregation: The endophenotype is more prevalent among the ill relatives of ill probands compared with the well relatives of the ill probands.
Biological and clinical plausibility: The endophenotype bears some conceptual relationship to the disease.
We will present and discuss endophenotypes at two levels: First, we will dissect the DSM-IV phenotype into narrow psychopathological characteristics that are biologically and clinically meaningful and can be assessed quantitatively. We will refer to these core psychopathological features of major depression as key components; we will refer to those key components that meet some of the endophenotype criteria as psychopathological endophenotypes. Second, we present biological markers that are useful for genetic studies. We will refer to biological markers that meet some of the endophenotype criteria as biological endophenotypes. All endophenotypes presented in this paper have to be seen as putative endophenotypes because whether or not they consistently meet all required endophenotype criteria has not been determined yet. We will review the literature and indicate the strength and limitations of psychopathological and biological endophenotypes in MDD with a special emphasis on studies associating endophenotypes with genes. Finally, we present future directions for the elucidation of the genetic basis of MDD.
KEY COMPONENTS OF MAJOR DEPRESSION
The validity of symptoms and components of MDD has been difficult to achieve because the MDD criteria likely encompass a group of disorders that are heterogeneous with respect to etiology and pathophysiology. Based on factor and cluster analytic studies, Nelson and Charney (1981) found that mood bias toward negative emotions, anhedonia, and psychomotor symptoms best characterize MDD. However, the diagnostic criteria for a disorder based on clinical characteristics without knowledge of the underlying etiological processes cannot be validated adequately. The use of more indirect indicators of disease validity or validity of depressive subtypes such as family history, treatment response, longitudinal course and stability, patterns of comorbidity, and social consequences led to circularity in validating the criteria (Kendell, 1989). Not surprisingly, studies on the biological basis of depression have found stronger associations between specific biological dysfunctions and certain components of major depression than with the presence or absence of DSM-IV MDD: symptoms such as cognitive deficits, rumination, psychomotor retardation, anhedonia, and lowered mood have been associated with specific focal abnormalities of regional cerebral blood flow (CBF; Mayberg et al, 1999; Drevets, 2000). Moreover, components of major depression such as altered response to stress (Caspi et al, 2003), impaired cognitive abilities, and dysfunctional reward-related behaviors are easier to model in animals than the depressive syndrome itself (Redei et al, 2001). Finally, intermediate levels of recurrence of depressive episodes have been associated with high genetic liability of MDD (Kendler et al, 1999), while a high temporal stability of the phenotype is favorable for genetic studies. The ensuing discussion dissects MDD into its key components based on biological validity, availability of accurate and quantitative assessment methods, and clinical relevance.
Depressed Mood (Mood Bias Toward Negative Emotions)
Depressed mood is a core symptom of MDD (Nelson and Charney, 1981). Attentional and mnemonic biases toward processing of mood-congruent information including sad, unpleasant and negative words, emotional facial expressions, and memories also are reliable and relatively specific neuropsychological findings of MDD (Watkins et al, 1996; Murphy et al, 1999), and have been found in remitted depressed subjects suggesting some state-independence (Hammen et al, 1985; Koschack et al, 2003). Experimental depletion of central serotonin led to the emergence of mood-congruent memory bias (Klaassen et al, 2002), and physiological activation of the brain regions that have been found to be involved in mood-congruent information processing (Elliott et al, 2002) is thought to play a key role in MDD (Drevets et al, 1997), thus implicating good biological validity. Attentional and mnemonic biases in depression have high clinical relevance because they are related to cognitive-behavioral theories of depression, upon which treatment strategies have been based (Beck, 1967). However, there is only preliminary data on the heritability of mood bias: recurrent thoughts of death and suicide appeared to be a specific characteristic of familial MDD (Kendler et al, 1999).
Anhedonia (Impaired Reward Function)
Loss of interest, lack of reactivity, and anhedonia also constitute core features of major depression (Nelson and Charney, 1981), and these symptoms represent key diagnostic criteria for the DSM-IV melancholic subtype of major depression. Anhedonia seems to be a relatively specific feature of depression (Fawcett et al, 1983), and even in patients with schizophrenia, anhedonia has been related to the depressive syndrome rather than to the deficit syndrome (Loas et al, 1999). Associations between dysfunctions of the brain reward system and anhedonia are the basis of the biological plausibility of anhedonia-related endophenotypes. Specifically, neurotrophic factors including cAMP response element binding protein (CREB) and brain-derived neurotrophic factor (BDNF) and the transcription factor delta-FosB may represent molecular mechanisms involved in long-term alterations of the brain reward system (Nestler et al, 2001; 2002). Enhanced rewarding effects of dextroamphetamine found in patients with MDD may represent hypofunction of the dopaminergic system associated with anhedonia (Tremblay et al, 2002). Preliminary evidence for the potential heritability of these findings includes a functional polymorphism of the catechol-O-methyltransferase (COMT) gene that has been associated with the individual variation in the brain response to dopaminergic challenge (Mattay et al, 2003).
Epidemiological research provides clues for state-independence, heritability, and familial association of dysfunctions of the brain reward system as endophenotype for MDD. For example, anhedonia symptoms often precede the onset of MDD (Dryman and Eaton, 1991) and seem to be relatively stable over time (Oquendo et al, 2004). A sib-pair study showed that the personality construct ‘reward dependence’ has trait-like characteristics related to the familiality of major depression (Farmer et al, 2003). Given the extensive evidence for associations between impairments of brain reward pathways and addiction (Nestler et al, 2002), familial coaggregation and lifetime comorbidity of substance use disorders and MDD (Kendler et al, 1997; Brook et al, 2002) also suggest persistent familial abnormalities of brain reward pathways in MDD.
Impaired Learning and Memory
Diminished ability to attend or concentrate is a diagnostic criterion of DSM-IV major depressive episode, which reflects cognitive impairments including learning and long-term memory, and deficits in working memory (short-term memory) and selective attention (Burt et al, 1995; Landro et al, 2001). These impairments are not specific for depression, although the pathophysiological mechanisms underlying these features of depression may differ from those in other psychiatric disorders (Berman et al, 1993; Barch et al, 2003). Specifically, abnormal reduction of CBF and metabolism in the dorsolateral prefrontal cortex and dorsal anterior cingulate cortex (ACC) might explain diminished ability to attend and concentrate in MDD (Drevets and Raichle, 1998), thus providing some evidence for the biological plausibility of this putative endophenotype for MDD.
Impaired concentration and attention were found to be frequent and early prodromal signs of depression (Dryman and Eaton, 1991; Hagerty et al, 1997), and recovered patients sometimes continue to show memory impairment (Roiser et al, 2003). However, at the symptom level, these deficits have been found to be relatively unstable over the course of depressive illness (Oquendo et al, 2004). Some authors suggest that working memory deficits in depression are due to persistent deficits in selective attention (state-independence) assessed by the Stroop paradigm that have been related to persistent abnormalities in the prefrontal cortex (Trichard et al, 1995; Blumberg et al, 2003). In contrast, deficits in long-term storage and retrieval of declarative memory have been associated with number of depressive episodes, stress, hypercortisolemia, and hippocampal volume reduction found in major depression (MacQueen et al, 2002; 2003), thus suggesting that these memory symptoms are rather a consequence than an etiologic factor of depression.
There is a wealth of knowledge on the genetics of working memory in mice providing converging evidence that glutamate, acetylcholine and dopamine receptors, calcium/calmodulin kinase II and protein kinase C gene families play roles in short-term information processing (Luciano et al, 2003). In healthy humans and schizophrenic patients, a functional polymorphism of the COMT gene appeared to explain some of the individual variance in working memory performance (Egan et al, 2001; Malhotra et al, 2002). However, there is a lack of information on the heritability, familial association, and cosegregation of learning and memory symptoms related to MDD.
Neurovegetative Signs
Although appetite and weight change are not specific symptoms of major depression (Nelson and Charney, 1981), there is some evidence that the direction of appetite and weight change may be useful as a marker of depressive subtypes. The direction and extent of weight change was found to be consistent across episodes of MDD (Stunkard et al, 1990) suggesting some state-independence, although a recent study did not show any correlation between Hamilton depression scale appetite measures across episodes (Oquendo et al, 2004). Evidence for heritability and familial association comes from studies in identical twins showing that melancholic and atypical depressive subtypes independently aggregate in families and are at least in part genetically determined, and that these subtypes are most clearly distinguished by a different pattern of appetite and weight change (Kendler et al, 1996). Finally, appetite-associated endophenotypes for depression seem to be biologically plausible given that brain monoamines and peptides that play major roles in major depression, such as serotonin, norepinephrine, dopamine, neuropeptide Y, and corticotropin-releasing hormone, also play important roles in the regulation of food intake and body weight (Gillard et al, 1993; Meguid et al, 2000; Wang et al, 2001).
More than 90% of depressed patients complain about impairments of sleep quality, and insomnia was found to be a prominent risk factor for subsequent development of depression. However, sleep disturbances including morning awakenings are not specific for major depression and co-occur with a wide range of physical and psychological symptoms. In addition, studies comparing subtypes of depression such as unipolar, melancholic, and bipolar depression failed to find consistent differences in nocturnal sleep patterns (Riemann et al, 2001). However, due to the availability of sophisticated examination methods such as polysomnography, sleep pathophysiology may be well suited to discover endophenotypes for major depression (see below).
Diurnal Variation
Some symptoms of MDD may show diurnal variations (mood, psychomotor activity, accessibility of memories of positive and negative experiences), and a subgroup of patients with MDD may have a circadian rhythm disorder (Bunney and Bunney, 2000). In healthy young subjects, moderate changes in the timing of the sleep–wake cycle had specific effects on subsequent mood (Boivin et al, 1997). The association between phase advance of the sleep–wake cycle and phase advances in nocturnal cortisol secretion, and the effect of antidepressants on circadian rhythms of behavior, physiology, and endocrinology contribute to the biological plausibility of this putative endophenotype (Duncan, 1996; Bunney and Bunney, 2000). Because manipulations of the circadian rhythms (light therapy, sleep deprivation, phase advance treatment) can have antidepressant efficacy, circadian abnormalities have been hypothesized to be etiologically associated with major depression.
There is strong evidence for a genetic control of the human circadian clock (Linkowski et al, 1993), and a familial variant of human sleep behavior has been attributed to a mutation in a human clock gene (Toh et al, 2001). The influence of a functional polymorphism within the angiotensin I-converting enzyme (ACE) gene on partial sleep deprivation in patients with MDD is probably mediated by dopaminergic neurotransmission (Baghai et al, 2003). A functional polymorphism within the promoter of the serotonin transporter gene has been related to sleep deprivation's efficacy and suggests an involvement of the central serotonergic system in the regulation of circadian rhythms (Benedetti et al, 1999).
Further epidemiological research is needed to determine state-independence, heritability, and familial aggregation of circadian abnormalities associated with MDD.
Impaired Executive Cognitive Function (Response Speed)
Impairments of executive cognitive function in depressed subjects refer to abnormalities in cognitive behaviors that control and integrate neural activities including selecting strategies, planning, and monitoring performance. These impairments are not specific for MDD and usually recover to normal levels in remission. However, response speed has been found to be unrelated to concurrent depressive symptoms and to remain impaired in fully recovered depressed patients off medication (Roiser et al, 2003) (state-independence). Specifically, inspection time, a measure of speed of information processing that does not require a speeded motor response, has been found to be slower in subjects with unipolar major depression than in age-, sex-, and IQ-matched controls independent of current mood (Tsourtos et al, 2002). The cholinergic basis of inspection time (Nathan and Stough, 2001) paralleling the hypothesis of a cholinergic dysfunction in major depression (Riemann et al, 1994) lends biological plausibility to this putative endophenotype. Twin studies demonstrated heritability for inspection time, sharing a substantial genetic relationship with performance IQ (Luciano et al, 2003).
Psychomotor Change (Retardation, Agitation)
Psychomotor retardation and agitation are included in the DSM-IV diagnostic criteria of major depression and have been shown to reliably differentiate depressed patients from psychiatric and normal comparison groups (Sobin and Sackeim, 1997). Moreover, psychomotor disturbance has been proposed as a marker of an underlying neuropathological process specific for the melancholic depressive subtype (Parker, 2000). The biological and clinical plausibility for this putative endophenotype includes associations between psychomotor disturbances and hypothalamic-pituitary-adrenocortical (HPA) axis dysfunction in depressed subjects (Mitchell et al, 1996), and Parkinsonian movement deficits in melancholic patients (Rogers et al, 2000), consistent with a potential dopaminergic dysfunction related to depression (Nestler et al, 2002). Unfortunately, information on state-independence, heritability, familial association, and cosegregation of specific psychomotor disturbances are lacking.
Increased Stress Sensitivity (Gender Specific)
The heritability of major depression being estimated to range between 31 and 42% emphasizes the relative importance of environmental factors (Sullivan et al, 2000). Given the lack of specificity between stressors and psychopathological outcomes (McMahon et al, 2003), one may hypothesize that gene–stressor interactions account for outcome specificity. Therefore, psychopathological constructs reflecting gene–environment interactions might be among the most specific and most useful endophenotypes for major depression. Caspi et al (2003) have shown in a representative prospective study that 5-HTT genotypes moderate the influence of stressful life events on major depression, thus successfully using a gene–environment interaction as phenotype.
Gender differences have appeared as a specific characteristic of stress sensitivity in humans. Men and women are, in general, equally sensitive to the depressogenic effects of stressful life events, but their responses vary depending upon the nature of the event itself. Men are more likely to have depressive episodes following divorce, separation, and work difficulties, whereas women are more sensitive to events in their proximal social network, such as difficulty getting along with an individual, serious illness, or death (Kendler et al, 2001).
Diathesis-stress theories of depression predict that genes influence individuals' sensitivity to stressful events (Costello et al, 2002), consistent with a potentially important role of gene-by-environment interactions played in the etiology of depressive psychopathology. A diagnostic construct consistent with these theories is the personality concept ‘neuroticism’ defined as general vulnerability to anxiety and depressive symptoms under stress. Neuroticism was found to be reasonably invariant during adulthood (Costa and McCrae, 1988), although depressive state features may have a considerable impact on neuroticism scores (Griens et al, 2002). There is consistent evidence of gender-specific heritability and familial association and cosegregation for neuroticism as putative endophenotype for MDD: neuroticism has been associated with gender-specific genetic factors, and a high degree of overlap between genes influencing neuroticism and major depression within the genders has been demonstrated (Fanous et al, 2002); the reported heritability of neuroticism is equal or greater than heritability estimates for MDD (Jang et al, 1996); and neuroticism has been successfully used as phenotype for genetic studies (Sen et al, 2003).
The biological plausibility of stress-related endophenotypes for MDD can be derived from biological correlates of increased stress sensitivity including an excessive activation of the HPA axis that is frequently found in patients with MDD (see below). Sex differences in the response of the HPA axis to stress appeared to be important: overall, women showed a greater stress responsiveness than men, consistent with the greater incidence of major depression in women (Young, 1998); moreover, men showed greater cortisol responses to achievement challenges, whereas women showed greater cortisol responses to social rejection challenges (Stroud et al, 2002).
Although the concept of gene–environment interactions may be most closely related to the concept of major depression, psychometric difficulties, and the complexity and temporal structure of its underlying molecular mechanism complicate its practical use. It is particularly noteworthy that recent studies have demonstrated nongenomic transmission across generations of not only maternal behavior but also stress responses (Francis and Meaney, 1999). This has clear parallels in clinical population. For example, environmental events (for example, early childhood stressors) correlate with the development of psychiatric disorders in adults (Heim and Nemeroff, 2001). Indeed, as witnessed by multiple studies of discordant monozygotic twins where one has the disorder and the other does not, epigenetic mechanisms must be operative to control behavior in genetically identical populations (Petronis et al, 2003). A critical question involves the mechanism by which early life events regulate long-term changes in behavior and sustained differences in gene expression.
In most areas of the brain, neurons are not replaced; thus, permanent and semipermanent modifications that occur in early life, which alter gene transcription, could have downstream effects that may be temporally distant from the initial event. These nongenetic (epigenetic) mechanisms of gene regulation likely play a role in the formation of cellular memory and the modulation of neural circuitry in a manner that alters lifetime cellular and behavioral responses in an organism. The true extent of the dynamic mechanisms responsible is unknown and is an active area of research. However, it is known to involve the interplay of transcription factors interacting with covalent DNA modifications, such as cytosine methylation, and the accessibility of DNA that is regulated by histone acetylation (Petronis, 2001; Geiman and Robertson, 2002). These mechanisms are likely involved in modulating how previous experience may regulate subsequent behavioral responses.
BIOLOGICAL ENDOPHENOTYPES
The number of genes involved in a phenotype is thought to be associated with both the complexity of the phenotype and the difficulty of the genetic analysis (Gottesman and Gould, 2003). Assuming a large number of genes involved in the pathogenesis of major depression, and assuming that disease progression and medication may alter the clinical phenotype, defining endophenotypes for depression by narrow psychopathological definitions such as circadian abnormalities and direction of vegetative symptoms, by personality constructs such as neuroticism and reward dependence, or by neuropsychological tests including inspection time and the Stroop paradigm may still require relatively large samples to detect the effect of a single genetic polymorphism. One emerging strategy that may circumvent some of these difficulties is the use of quantitative biological markers, which are associated with disease and may be closer to single gene effects than clinical phenotypes (Almasy and Blangero, 2001). The ideal biological endophenotype would be a biological marker that is easy to assess and that reflect the action of a single gene.
The use of biological endophenotypes has been successful in locating genes for nonpsychiatric disorders. For example, some of the multiple genes involved in cardiac arrhythmias were identified using an ECG endophenotype. Initially, a prolonged QT interval on the electrocardiogram was found in some patients with familial cardiac arrhythmia. Subsequent discovery of an increased prevalence of a prolonged QT interval among healthy relatives of arrhythmic patients than among unrelated controls suggested heritability of this ECG trait in these families. Finally, using QT interval elongation as endophenotype allowed for successful genetic linkage studies (Keating et al, 1991; Keating and Sanguinetti, 2001).
In the following, we will present biological correlates for major depression that meet some of the endophenotype criteria.
REM Sleep
Reduced REM latency, higher REM density, and more REM sleep in adolescents turned out to be specific and predictive for unipolar major depression, whereas none of these signs were found in adolescents who later switched to bipolar disorder and in those who remained free from psychopathology at follow-up (Rao et al, 2002). Early changes of REM sleep parameters, especially REM latency and percentage amount of REM during the sleep period, have been found in patients with acute and remitted major depression (Giles et al, 1993) (state-independence). The first-degree relatives of unipolar depressed patients showed a significantly higher REM density than controls, suggesting that genetic factors contribute to these REM sleep alterations (Giles et al, 1998; Modell et al, 2003). Nearly all effective antidepressant medications have shown a pronounced inhibition of REM sleep (Murck et al, 2003), and the serotonergic and cholinergic neurotransmitter systems have been implicated in both REM sleep regulation and MDD, thus providing evidence for the clinical and biological plausibility of this putative endophenotype.
Several candidate genes may be considered for REM sleep-related endophenotypes. The CREB gene has been implicated in the regulation of REM sleep (Graves et al, 2003), memory consolidation, and major depression (Zubenko et al, 2002). The effect of the muscarinic cholinergic 2 receptor gene that has been implicated in MDD in women (Comings et al, 2002) may conceivably be mediated by REM sleep disturbances. Finally, genes associated with neurological disorders affecting REM sleep characteristics such as narcolepsy may contribute to REM sleep disturbances in MDD: the HLA class II allele DQB1*0602, genes for the hypocretin receptors, and polymorphisms in genes of the COMT and the tumor necrosis factor system (Taheri and Mignot, 2002).
Abnormalities in Brain Structure and Function
The identification of pathologic lesions in specific regions of the central nervous system has importantly contributed to the rapid progress in the understanding and treatment of neurological disorders including Parkinson's and Alzheimer's disease. Neuropathological findings are extremely useful to define nosological subtypes reflecting different underlying disease states that may be related to genetic vulnerability factors. For example, clinical features alone can only be used to diagnose parkinsonism; postmortem examination is needed for the definite diagnosis of the underlying disease including classic Parkinson's disease, multiple system atrophy, progressive supranuclear palsy, and frontotemporal dementia (Giasson and Lee, 2003).
Although there is no consensus in the field about the site of pathology in major depression, functional, structural, and molecular brain mapping in major depression have the potential to bridge the gap between clinical depressive features and genes. Since approximately 70% of all genes are expressed in the brain, many functional gene polymorphisms can potentially influence how the brain processes information. Since functional imaging has the capacity to assay within individuals information processing in discreet brain circuits, it has the potential to provide endophenotypes for MDD (Hariri and Weinberger, 2003). For example, significant differences have been reported between s/s and l/l genotypes of the 5-HTT promoter gene regarding the amygdala response to fearful faces measured by functional MRI in the absence of behavioral differences (Hariri et al, 2002). Furthermore, growing evidence suggests that major depression travels with brain structural changes mediated by hypercortisolemia, glutamate neurotoxicity, stress-induced reduction in neurotrophic factors, and stress-induced reduction in neurogenesis (Sheline, 2003). Therefore, advanced imaging technology might describe subtle changes in brain structures that are associated with specific pathophysiological processes and genes. Finally, in vivo molecular imaging opens up the potential to connect findings in genetic neuroscience obtained from animal experiments and postmortem human studies to clinical characteristics of depressed subjects by defining endophenotypes at the molecular level (eg receptors, transporters, and enzymes).
Functional imaging
A series of studies have consistently found that the resting CBF and glucose metabolism in the amygdala in subjects with familial pure depressive disease and the melancholic depressive subtype is increased (Drevets, 2000). Elevation of resting amygdala CBF and metabolism may prove specific to primary mood disorders, insofar as this abnormality has not been reported in obsessive–compulsive disorder, panic disorder, phobic disorders, schizophrenia, or other neuropsychiatric conditions (Charney and Drevets, 2002). Although the magnitude of the amygdala activity in depression is partly modulated by illness severity, preliminary data suggest that left amygdala activity is abnormally elevated in remitted depressed patients off medication with a family history for affective disorders (Drevets et al, 1992). Moreover, one study showed that increased amygdala activity predicted return of depressive symptoms in remitted MDD subjects on medication following tryptophan depletion (TD; Bremner et al, 1997). The associations between elevated amygdala activity and vegetative depressive symptoms, plasma cortisol (Drevets et al, 1992), and REM sleep (Nofzinger et al, 1999) underline the plausibility of this biological marker for MDD. However, whether elevated amygdala activity meets the endophenotype criteria heritability, familial association, and cosegregation has not yet been determined.
In the subgenual prefrontal cortex, decreased CBF and metabolism have been consistently implicated by numerous studies in MDD (Drevets, 2000; Kegeles et al, 2003). Dysfunction of this brain region has been associated with blunted hedonic response and exaggerated stress responsiveness (Pizzagalli et al, 2004), and functional alterations in this region have been observed following serotonergic challenge (Kegeles et al, 2003), induced sadness (Mayberg et al, 1999), and treatment with a serotonin reuptake inhibitor (Buchsbaum et al, 1997). In remitted depressed patients, mood provocation produced a CBF decrease in brain regions connected with the subgenual prefrontal cortex (Liotti et al, 2002). Although information about this finding in healthy subjects at risk for MDD is lacking, it represents another promising imaging endophenotype.
Structural imaging
Volume reductions in the ACC located ventrally (‘subgenual’) and anterior (‘pregenual’) to the genu of the corpus callosum have been implicated by numerous studies of mood disorders (Drevets, 2001). Specifically, a volume reduction in the left subgenual ACC has been associated with familial affective disorders by MRI-morphometric measures (Drevets et al, 1997; Hirayasu et al, 1999) and by postmortem neuropathological studies, which have shown glial reduction in the corresponding gray matter (Öngür et al, 1998). This reduction in volume exists early in the illness (state-independence), but appears to become more pronounced following illness onset, based upon preliminary evidence in twins discordant for MDD (Botteron et al, 1999). Humans with lesions that include the ventral ACC show abnormal autonomic responses to emotional stimuli, an inability to experience emotion related to concepts, and inability to use information regarding the probability of aversive social consequences vs reward in guiding social behavior (Damasio et al, 1990). In rats, left-sided lesions of the ACC increase sympathetic arousal and corticosterone responses to restraint stress (Sullivan and Gratton, 1999). The mechanisms and genes underlying volume loss in the ACC have not yet been determined. Preclinical studies on the role and genetics of neurotrophic factors and the signaling cascade neurotrophic factor/MAP kinase/bcl-2 involved in the fine balance maintained between the levels and activities of cell survival and cell death factors (Manji et al, 2003b) may inform clinical studies associating ACC volume loss to genes.
Reductions in hippocampal volumes associated with MDD have been consistently reported. However, this structural abnormality is not a specific sign of MDD (ie it has been found in patients with PTSD, schizophrenia, and epilepsy) and only occurs in a subgroup of MDD patients (Sheline and Mintun, 2002). In some studies, the volume loss appears to have functional significance with an association between acute depression and abnormalities of declarative memory (Burt et al, 1995) as well as associations between depression in remission and lower scores on tests of verbal memory (Sheline et al, 1999). The pathogenesis of hippocampal volume reduction seems to overlap with depressive pathophysiology, given its association with early-life stress, stress hormones, and duration of depressive illness (McEwen, 1999; Brunson et al, 2001; MacQueen et al, 2003). A study in monkeys suggests that hippocampal volume also reflects an inherited characteristic of the brain associated with increased cortisol response to stress (Lyons et al, 2001). Preliminary results on the genetics of the hippocampal function suggest that the BDNF val66met polymorphism may influence the development of memory deficits associated with psychopathology (Egan et al, 2003), and targeted disruption of mineralocorticoid receptor (MR) genes resulted in impaired hippocampal neurogenesis (Gass et al, 2000).
The literature is in disagreement regarding amygdala volumes in mood disorders, possibly due to technical limitations such as low reliability and validity of amygdala volume measures. However, a recent study using high-resolution 3T MRI has consistently found decreased amygdala volume in symptomatic and remitted mood disorders (Drevets et al, 2004). Amygdala volumes have been negatively associated with amygdala activity, suggesting that amygdala hyperactivity could be a factor in amygdala volume reductions in MDD (Siegle et al, 2003). This association and the high clinical plausibility of neuropathological abnormalities of the amygdala as endophenotype for MDD encourages further investigation on the consistency, specificity, familiality, and heritability of these findings.
Receptor pharmacology
Decreased 5-HT1A receptor binding potential has been consistently found in multiple brain areas of patients with MDD (Drevets et al, 1999; 2000). This abnormality is not highly specific for MDD and has been found in patients with panic disorder (Neumeister et al, 2004) and temporal lobe epilepsy (Toczek et al, 2003), and may explain the considerable comorbidity among these conditions. The lack of effect of selective serotonin reuptake inhibitor treatment and hydrocortisone challenge on 5-HT1A receptors in recovered patients with MDD suggests state-independence of this abnormality (Bhagwagar et al, 2003; 2004). Unfortunately, no information is available on the heritability, familial association, and cosegregation of 5-HT1A receptor binding potential. However, a recent report suggests that a polymorphism associated with 5-HT1A receptor transcription is more common in MDD than in controls (Lemonde et al, 2003).
There is growing evidence from animal studies for the biological plausibility of this putative endophenotype for stress-related disorders. Mice with mutation in the 5-HT1A receptor gene have been consistently found to display increased stress-like behaviors and to express decreased activity toward a novel object (Bakshi and Kalin, 2002). However, the use of a tissue-specific, conditional rescue strategy revealed that the expression of the 5-HT1A receptor in the hippocampus and cortex (but not in the raphe nuclei) during the early postnatal period (but not in the adult) is sufficient to rescue the normal behavioral phenotype of the knockout mice (Gross et al, 2002), suggesting that stimulation of the 5-HT1A receptor during the postnatal period leads to long-lasting changes in the brain structure that are necessary for normal affective behavior throughout life.
Serotonin, Dopamine, and Norepinephrine
Alterations in noradrenergic and serotonergic function in the brain have been implicated in the pathophysiology of depression and the mechanism of action of antidepressant drugs (Charney, 1998), although dysfunctions within the monoaminergic neurotransmitter systems are not likely to play primary roles in the pathophysiology of depression but rather represent the downstream effects of other, more primary abnormalities (Manji et al, 2003b). Because depletion of catecholamines or serotonin induces significant depressive symptoms in remitted depressed subjects, depressive reactions in response to lowering of brain monoamine neurotransmitters has been proposed as an endophenotypic vulnerability marker for major depression (Berman et al, 1999).
Serotonin
Major depression has been associated with abnormally reduced function of central serotonergic systems by abundant evidence from biochemical, challenge, imaging, and postmortem studies (Coppen et al, 1973; Charney et al, 1981; Lopez et al, 1998; Drevets et al, 1999). TD is an instructive paradigm for investigating the relationship between serotonergic function and depression. This paradigm involves the mood response to serotonin depletion, achieved by oral loading with all essential amino acids except the 5-HT precursor, tryptophan. The transient reduction in plasma tryptophan concentrations, cerebral serotonin synthesis, and central 5-HT concentrations resulting from this dietary manipulation is associated with redevelopment of depressive symptoms in remitted depressed patients who are either off medication (Delgado et al, 1994) or medicated with selective serotonin reuptake inhibitors (Delgado et al, 1999).
TD-induced depressive symptoms show a relatively high specificity for major depression (Bell et al, 2001). The presence of this diagnostic sign in remitted patients suggests state-independence. TD-induced depressive symptoms seem to be heritable: in remitted depressed patients, the long allele of the serotonin transporter gene promoter polymorphism predicted depressive response to TD (Moreno et al, 2002), while, in healthy women, the s-allele of this functional polymorphism and a positive family history of depression represented additive risk factors for TD-induced depressive symptoms (Neumeister et al, 2002). There is also evidence for familial association and cosegregation: never-depressed subjects with a positive family history of depression have also been shown to experience mood symptoms following TD that were smaller than in remitted depressed patients but different from subjects without familial risk who showed no effect following TD (Benkelfat et al, 1994).
In vulnerable individuals, TD induces mood-congruent memory bias (Klaassen et al, 2002), alters reward-related behaviors (Rogers et al, 2003), impairs memory consolidation (Riedel et al, 2002), slows responses to positive stimuli, and disrupts inhibitory affective processing (Murphy et al, 2002). These TD-induced changes are comparable with those occurring in major depressive episodes, thus implicating clinical plausibility. Acute severe serotonin depletion leads to biological changes associated with MDD, including enhanced norepinephrine transporter mRNA levels and reduced serotonin transporter mRNA levels (Koed and Linnet, 2000), increased number of MR binding sites (Semont et al, 1999), and altered BDNF gene expression in the dentate gyrus (Zetterstrom et al, 1999), thus implicating biological plausibility.
Norepinephrine/dopamine
MDD has been associated with noradrenergic and dopaminergic dysfunction. Findings implicating catecholaminergic dysfunction in the pathophysiology of depression include studies about neurotransmitter synthesis and neurotransmitter storage, showing that reduction of catecholamine stores results in an exacerbation of depressive symptoms (Mendels and Frazer, 1974).
An instructive paradigm for investigating the relationship between catecholaminergic function and depression has involved the mood response to catecholamine depletion (CD), achieved by the administration of the tyrosine hydroxylase inhibitor α-methylparatyrosine (AMPT). Mood responses to CD in healthy subjects are usually not significant (Salomon et al, 1997). Among untreated, symptomatic depressed patients prior to initiation of an antidepressant treatment CD failed to exacerbate depression (Miller et al, 1996b). This finding may be due to the brain catecholamine function being already maximally dysfunctional in symptomatic depressed patients (ceiling effect) (Lambert et al, 2000). Among treated depressed patients, CD reversed the antidepressant effects of antidepressants, in particular of catecholamine reuptake inhibitors (Miller et al, 1996a) and light therapy (Neumeister et al, 1998).
Because CD has not been systematically used in high-risk subjects and across different neuropsychiatric disorders, there is a lack of information on the specificity, familial association, and cosegregation of CD-induced symptoms for MDD. The presence of CD-induced depressive symptoms in unmedicated remitted patients with MDD suggests state-independence of this biological marker (Berman et al, 1999).
The depressive symptoms evoked by CD were often similar to those the patients had experienced during their depressive episode, suggesting clinical plausibility. There is also evidence for biological plausibility: the CD-induced return of depressive symptoms has been associated with decreased brain metabolism in orbitofrontal and dorsolateral prefrontal cortex; increased resting metabolism in prefrontal cortex and limbic areas have been found to increase vulnerability to CD-induced depressive exacerbation (Bremner et al, 2003).
HPA Axis and CRH
Altered HPA axis physiology and dysfunctions of the extrahypothalamic CRH system have been consistently found in humans with major depression. There is accumulating evidence that altered stress hormone secretions in depression are more than epiphenomenal, and that antidepressants may act through normalization of these changes. Given the potentially causal role of HPA axis and CRH system dysfunctions in depressive pathophysiology, and the involvement of cortisol and CRH in a variety of biological and behavioral components of major depression, indicators of these dysfunctions are likely to be useful as endophenotypes for major depression.
Cortisol
Negative feedback regulation of the HPA axis occurs through a dual-receptor system of MRs and glucocorticoid receptors (GRs). Decreased limbic GR receptor function (Modell et al, 1997; Mizoguchi et al, 2003) and increased functional activity of the MR system (Young et al, 2003) suggest an imbalance in the MR/GR ratio in stress-related conditions such as MDD. The possible antidepressant properties of GR antagonists also appear compatible with the corticoid receptor hypothesis (Belanoff et al, 2002).
The neuroendocrine function test that measures dysfunctional cortisol responses in major depression most sensitively combines the dexamethasone suppression test and the CRH stimulation test (dex/CRH test) (Heuser et al, 1994). The specificity of this test is high to discriminate between healthy subjects and psychiatric patients including patients with panic disorder, mania, and schizophrenia, and its sensitivity for MDD is above 80%, depending on age and gender; however, its specificity for MDD with regard to other psychiatric disorders is low (Heuser et al, 1994). Abnormal cortisol responses in MDD patients, MDD high-risk probands, and healthy controls were found to be surprisingly constant over time (Modell et al, 1998), and independent of the actual depressive state (Zobel et al, 1999), suggesting that this marker is state-independent. Findings in healthy subjects at high familial risk for affective disorders (Holsboer et al, 1995) suggest familial association and cosegregation.
Interactions between cortisol and its receptors and neurotransmitters, neuropeptides, and brain circuits associated with depressive symptomatology suggest biological plausibility of the dex/CRH test as endophenotype for MDD. Specifically, the MR system seems to control the sensitivity of the CRH-1 system (de Kloet, 2003), which is thought to be altered in MDD, and to be the primary mediator of 5-HT1A downregulation after chronic stress (Kuroda et al, 1994), whereas the GR seems to be the primary receptor involved in stress-related 5-HT2A receptor upregulation (Karten et al, 1999).
Depression-like alterations of functions of the prefrontal cortex such as inhibitory control, attention regulation, and planning following cortisol administration, and the bidirectional associations between amygdala activity and cortisol levels (Gold et al, 2002) suggest clinical plausibility of cortisol-related endophenotypes for MDD. Furthermore, elevated cortisol may mediate between major depression and its medical long-term consequences such as coronary heart disease, type II diabetes, and osteoporosis.
Finally, there is some preliminary evidence for the heritability of GR/MR system functions. Some humans manifest a relative glucocorticoid resistance caused by GR gene mutations (Koper et al, 1997). In mice, conditional GR overexpression has been suggested as a model for increased anxiety-related behavior not secondary to altered levels of stress hormones (Muller et al, 2002). Targeted disruption of MR genes resulted in impaired hippocampal neurogenesis (Gass et al, 2000). Genetic factors most probably act in concert with environmental factors: an increase in hippocampal MR levels has been shown after exposure to psychological stress (Gesing et al, 2001).
Corticotropine-releasing hormone
The evidence for the specificity, and clinical and biological plausibility of endophenotypes related to dysfunctions of the hypothalamic and extrahypothalamic CRH system for MDD is abundant: Levels of CRH in the CSF are elevated in some depressed subjects (Nemeroff et al, 1984), while the pituitary response to CRH appears appropriate given the high cortisol levels (Gold et al, 1986); the number of CRH-secreting neurons in limbic brain regions is increased (Raadsheer et al, 1994); the number of CRH binding sites in the frontal cortex is reduced, possibly as a compensatory response to increased CRH concentrations (Nemeroff et al, 1988); CRH produces a number of physiological and behavioral alterations that resemble the symptoms of major depression including decreased appetite, disrupted sleep, decreased libido, and psychomotor alterations (Nemeroff, 1996); and anxiety and depression scores have been reduced following CRH-1 receptor blockade (Zobel et al, 2000).
One of the strongest models of environmental regulation of the development of responses to stress is the postnatal handling research. The central CRH system is seen as the critical target for these environmental effects (Francis and Meaney, 1999). Postnatal maternal separation increases CRH gene expression in the paraventricular nucleus and alters systems involved in the regulation of the CRH system (eg the noradrenergic system); these effects may become permanent. Additionally to these environmental regulations, genetic factors have to be taken into account: reports on mouse mutants where CRH receptors were genetically inactivated suggested that CRH-R1 mediates anxiety-like behavior (Timpl et al, 1998); the CRH binding protein gene has been found to be involved in the vulnerability for MDD (Claes et al, 2003).
Although the CRH system is among the most promising to provide clinically and biologically plausible endophenotypes for MDD, few studies have investigated the temporal stability, heritability, familial association, and cosegregation of the CRH system in relation to major depression. One reason for the lack of such studies is the lack of PET ligands for CRH receptors, which can permit noninvasive assessments of CRH system dysfunction in vivo in humans.
Intracellular Signaling Molecules (a Perspective)
Neurotrophic factors
The prominent cognitive deficits and brain volumetric changes in depression give additional weight to the contention that severe mood disorders arise, at least in part, from impairments of cellular plasticity and resilience (Manji et al, 2001; Manji and Duman, 2001). Endogenous neurotrophic factors such as BDNF are necessary for growth, survival, and functioning of neurons. They increase cell survival by providing necessary trophic support for growth, and also by exerting inhibitory effects on cell death cascades. There is emerging evidence, primarily from postmortem studies, that supports a role for abnormalities in neurotrophic signaling pathways in depression. Decreased levels of CREB, BDNF, and the TrkB receptor have been reported in suicide victims (Odagaki et al, 2001; Dwivedi et al, 2003).
As discussed already, genetic abnormalities in CREB and BDNF may also occur in depression. Substantial evidence indicates that CREB is a core component of the molecular switch that converts short- to long-term memory. A growing body of evidence suggests that antidepressants may regulate neurotrophic signaling cascades. Antidepressant treatment in rats increases CREB phosphorylation and CREB-mediated gene expression in mice limbic brain regions. More evidence that relates upregulation of these pathways and antidepressant utilization comes from antidepressant-like performance in behavioral models. Thus, it was observed that CREB overexpression in the dentate gyrus or BDNF injection results in an antidepressant-like effect in the learned–helplessness paradigm and the forced swim test model of antidepressant efficacy in rats (Chen et al, 2001; Shirayama et al, 2002). Together, the data suggest that alterations of neurotrophic signaling cascades may underlie the pathophysiology and treatment of depression. Because of the potential genetic underpinnings of abnormalities of these neurotrophic pathways, they may provide biological endophenotypes for major depression.
Ubiquitous signaling cascades
It is now clear that genetic abnormalities in signaling components are often fully compatible with life, and in many instances, despite the often-ubiquitous expression of the signaling protein, these abnormalities are associated with circumscribed clinical manifestations (Manji et al, 2003a). These overt, yet relatively circumscribed, clinical manifestations are believed to ultimately arise from vastly different transcriptomes (all of the transcripts present at a particular time) in different tissues because of tissue-specific expression, haploinsufficiency, genetic imprinting, alternate splicing, varying stoichiometries of the relevant signaling partners in different tissues, and differences in the ability of diverse cell types to compensate for the abnormality (either autocrine or paracrine). Moreover, there is preliminary evidence that abnormalities in shared signaling cascades may even play a role in the growing appreciation that ‘comorbidities’ of major depression including cardiac disease, migraines, atopic disease, type II diabetes, and anxiety disorders appear to be the rule rather than the exception. There is no question that some medical illnesses (eg osteoporosis) represent the secondary sequellae of the biochemical changes (eg hypercortisolemia, sympathetic hyperarousal) of depression; however, some comorbidities may arise because the signaling cascade (eg cAMP cascade) also plays a role in the pathophysiology of these other disorders (eg vascular reactivity in migraine). In support of such a contention, Bondy et al (2002) investigated the ACE ID and the G-protein beta3-subunit (Gbeta3) C825T polymorphism in 201 patients with major depression and 161 ethnically and age-matched controls. Both gene variants have earlier been implicated in cardiovascular disease or affective disorders, making them good candidates for a combined analysis. Analyzing the data for both genes, they found that the combined actions of ACE and Gbeta3 genotypes accumulate in carriers of the ACE D allele (ID and DD) and Gbeta3 TT homozygotes, with ID/DD-TT carriers showing a more than five-fold increase in risk for major depression. These intriguing findings suggest that heritable biological makers of common medical conditions may turn out to be useful endophenotypes for major depression.
CONCLUSIONS
Table 1 shows an overview of putative psychopathological and biological endophenotypes for MDD, indicating estimates of evidence for each putative endophenotype with respect to the endophenotype criteria (Tsuang et al, 1993).
Anhedonia-related endophenotypes showed good evidence regarding endophenotype criteria including specificity. These endophenotypes parallel Klein's ‘endogenomorphic’ depressive subtype reflecting ‘inhibited pleasure mechanisms’ as psychopathological core feature (Klein, 1974). However, longitudinal and high-risk studies using specific anhedonia measures and tasks that estimate abnormalities of the brain reward system are necessary to further evaluate the applicability of anhedonia-related endophenotypes for major depression.
Increased stress sensitivity also met most of the endophenotype criteria. Particularly, the familial coaggregation with MDD makes this phenotypic construct qualified for genetic studies; however, its use is limited by low specificity for MDD and by psychometric issues: the genetics of the partly state-dependent construct ‘neuroticism’, which can easily be assessed by a questionnaire, may be as complex as the genetics of MDD, and a potentially more reliable and more valid assessment of stress sensitivity by measuring stress levels and stress symptoms over time by means of a prospective community-based study is extremely costly and time-consuming.
Among the biological endophenotypes, TD showed evidence for all of the endophenotype criteria including specificity, state-independence, and familial association, thus encouraging the use of TD to identify a potentially homogenous depressive subtype associated with serotonergic dysfunction. Unfortunately, the complexity of TD including the use of a pharmacological challenge, the clinical observation of the research subject over many hours, and the exclusion of patients with symptomatic depression reduce the usefulness of this endophenotype, particularly in epidemiological research. The dex/CRH test, being somewhat easier to apply than TD, also showed good evidence across various endophenotype criteria. The low specificity for MDD, however, represents a major limitation to the use of this biological marker as MDD endophenotype.
The large sample sizes of MDD patients needed to perform genetic association studies on subsamples stratified according to endophenotypes is a general limitation to the endophenotype approach. In addition, using PET or fMRI endophenotypes may not be practical given the huge costs to obtain a sample size with sufficient power for genetic studies. The systematic evaluation of combinations of related psychopathological and biological endophenotypes (eg attentional bias toward negative stimuli combined with functional/structural abnormalities in the subgenual prefrontal cortex) expands the scope of this review.
FUTURE DIRECTIONS
Given the relative scarcity of well-designed twin, family, and prospective studies evaluating putative psychopathological and biological endophenotypes (see Table 1), future research has the potential to improve considerably the phenotypic definition of MDD. Progress in developing economical and easy-to-apply neurobiological markers may considerably facilitate the discovery of biological endophenotypes. Moreover, in the long term, the development of a new diagnostic system that includes psychopathological and biological endophenotypes will be necessary to improve the power of genetic studies by systematically defining relatively homogenous depressive subtypes.
The Framingham Heart Study, designed as a large community study with a long-term follow-up, helped to identify biological and behavioral risk factors for cardiovascular disease and to establish diagnostic criteria for nosological entities such as arterial hypertension. Likewise, prospective longitudinal studies that collect comprehensive phenotypic data of psychiatric disturbances (eg the Zurich Cohort study; Angst et al, 1984; Hasler et al, 2004) are required. These studies are very different from the conventional phenotypic data collection (DSM-disease present or absent); it is rather a systematic effort to quantify the manifestations that compose the overall phenotype. In addition, these long-term follow-up studies are also required to identify environmental modifiers and precipitants that may alter clinical and biological phenotypes. The definition of endophenotypes in a way that takes developmental and environmental factors into account to detect vulnerability genes (Caspi et al, 2002; 2003) is an exciting model for future epidemiological research in depression.
Neuroimaging, postmortem, and preclinical studies are required to discover neurobiological endophenotypes bridging the gap between behavioral phenotype and genotype. Specifically, postmortem studies on gene variation conducted in families or populations may prove very useful in studying many facets of the gene or genes in question. By linking gene and gene expression (mRNA) to brain structure and function, postmortem research can be used to identify genetically relevant disease subtypes and to validate neuroimaging findings. New PET ligands that have specificity for receptors implicated in the pathophysiology of major depression, which are sensitive to dynamic neurotransmitter function, are needed to associate baseline levels of receptor occupancy as well as receptor displacement in response to pharmacological and behavioral challenges to both behavioral phenotypes and genotypes. Dramatic progress in the development of MRI-based methods will help to reliably identify subtle abnormalities of neural structures, connectivity, and function in depressed subjects and healthy subjects at familial risk that may be used as endophenotypes for genetic studies (Charney et al, 2002). Finally, longitudinal neurobiological studies are required to elucidate impairments of neuroplasticity and cellular resilience in major depression (Manji and Duman, 2001).
Taken together, reducing phenotypic heterogeneity is crucial for the identification of vulnerability genes for major depression, and, therefore, the development of a new classification system is badly needed. We propose to dissect the behavioral phenotype into key components, and integrate specific environmental risk factors and neurobiological endophenotypes into the new classification system. We hope and expect that advances in epidemiology, neurobiology, and genetics will result in ongoing improvements of the phenotypic definition of major depression based on etiology and pathophysiology.
References
Almasy L, Blangero J (2001). Endophenotypes as quantitative risk factors for psychiatric disease: rationale and study design. Am J Med Genet 105: 42–44.
Angst J, Dobler-Mikola A (1984). The Zurich study. II. The continuum from normal to pathological depressive mood swings. Eur Arch Psychiatry Clin Neurosci 234: 21–29.
Angst J, Dobler-Mikola A. (1985). The Zurich study. VI. A continuum from depression to anxiety disorders? Eur Arch Psychiatry Clin Neurosci 235: 179–186.
Angst J, Dobler-Mikola A, Binder J (1984). The Zurich Study—a prospective epidemiological study of depressive, neurotic and psychosomatic syndromes. Eur Arch Psychiatry Clin Neurosci 234: 13–20.
Angst J, Merikangas KR (2001). Multi-dimensional criteria for the diagnosis of depression. J Affect Disord 62: 7–15.
Angst J, Sellaro R, Merikangas KR (2000). Depressive spectrum diagnoses. Compr Psychiatry 41: 39–47.
Angst J, Vollrath M, Merikangas K, Ernst C (1990). Comorbidity of anxiety and depression in the Zurich Cohort Study of Young Adults. In: Maser JD, Cloninger CR (eds). Comorbidity of Mood and Anxiety Disorders. American Psychiatric Press: Washington, DC. pp 123–137.
Baghai TC, Schule C, Zwanzger P, Zill P, Ella R, Eser D et al (2003). Influence of a functional polymorphism within the angiotensin I-converting enzyme gene on partial sleep deprivation in patients with major depression. Neurosci Lett 339: 223–226.
Bakshi VP, Kalin NH (2002). Animal models and endophenotypes of anxiety and stress disorders In: Davis KL, Charney DS, Coyle JT, Nemeroff CB (eds). Neuropsychopharmacology: The Fifth Generation of Progress. Lippincott Williams & Wilkins: Philadelphia, PA. pp 883–900.
Barch DM, Sheline YI, Csernansky JG, Snyder AZ (2003). Working memory and prefrontal cortex dysfunction: specificity to schizophrenia compared with major depression. Biol Psychiatry 53: 376–384.
Beck AT (1967). Depression: Clinical, Experimental and Theoretical Aspects. Harper & Row: New York.
Belanoff JK, Rothschild AJ, Cassidy F, DeBattista C, Baulieu EE, Schold C et al (2002). An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatry 52: 386–392.
Bell C, Abrams J, Nutt D (2001). Tryptophan depletion and its implications for psychiatry. Br J Psychiatry 178: 399–405.
Benedetti F, Serretti A, Colombo C, Campori E, Barbini B, di Bella D et al (1999). Influence of a functional polymorphism within the promoter of the serotonin transporter gene on the effects of total sleep deprivation in bipolar depression. Am J Psychiatry 156: 1450–1452.
Benkelfat C, Ellenbogen MA, Dean P, Palmour RM, Young SN (1994). Mood-lowering effect of tryptophan depletion. Enhanced susceptibility in young men at genetic risk for major affective disorders. Arch Gen Psychiatry 51: 687–697.
Berman KF, Doran AR, Pickar D, Weinberger DR (1993). Is the mechanism of prefrontal hypofunction in depression the same as in schizophrenia? Regional cerebral blood flow during cognitive activation. Br J Psychiatry 162: 183–192.
Berman RM, Narasimhan M, Miller HL, Anand A, Cappiello A, Oren DA et al (1999). Transient depressive relapse induced by catecholamine depletion: potential phenotypic vulnerability marker? Arch Gen Psychiatry 56: 395–403.
Bhagwagar Z, Montgomery AJ, Grasby PM, Cowen PJ (2003). Lack of effect of a single dose of hydrocortisone on serotonin(1A) receptors in recovered depressed patients measured by positron emission tomography with [(11)C]WAY-100635. Biol Psychiatry 54: 890–895.
Bhagwagar Z, Rabiner EA, Sargent PA, Grasby PM, Cowen PJ (2004). Persistent reduction in brain serotonin(1A) receptor binding in recovered depressed men measured by positron emission tomography with [(11)C]WAY-100635. Mol Psychiatry 9: 386–392.
Blumberg HP, Leung HC, Skudlarski P, Lacadie CM, Fredericks CA, Harris BC et al (2003). A functional magnetic resonance imaging study of bipolar disorder: state- and trait-related dysfunction in ventral prefrontal cortices. Arch Gen Psychiatry 60: 601–609.
Boivin DB, Czeisler CA, Dijk DJ, Duffy JF, Folkard S, Minors DS et al (1997). Complex interaction of the sleep–wake cycle and circadian phase modulates mood in healthy subjects. Arch Gen Psychiatry 54: 145–152.
Bondy B, Baghai TC, Zill P, Bottlender R, Jaeger M, Minov C et al (2002). Combined action of the ACE D- and the G-protein beta3 T-allele in major depression: a possible link to cardiovascular disease? Mol Psychiatry 7: 1120–1126.
Botteron KN, Raichle ME, Heath AC, Price A, Sternhell KE, Singer TM et al (1999). An epidemiological twin study of prefrontal neuromorphometry in early onset depression. Biol Psychiatry 45: 59S.
Bremner JD, Innis RB, Salomon RM, Staib LH, Ng CK, Miller HL et al (1997). Positron emission tomography measurement of cerebral metabolic correlates of tryptophan depletion-induced depressive relapse. Arch Gen Psychiatry 54: 364–374.
Bremner JD, Vythilingam M, Chin KN, Vermetten E, Nazeer A, Oren D et al (2003). Regional brain metabolic correlates of alpha-methylparatyrosine-induced depressive symptoms. JAMA 289: 3125–3134.
Brook DW, Brook JS, Zhang C, Cohen P, Whiteman M (2002). Drug use and the risk of major depressive disorder, alcohol dependence, and substance use disorders. Arch Gen Psychiatry 59: 1039–1044.
Brunson KL, Eghbal-Ahmadi M, Bender R, Chen Y, Baram TZ (2001). Long-term, progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin-releasing hormone reproduce the effects of early-life stress. Proc Natl Acad Sci USA 98: 8856–8861.
Buchsbaum MS, Wu J, Siegel BV, Hackett E, Trenary M, Abel L et al (1997). Effect of sertraline on regional metabolic rate in patients with affective disorder. Biol Psychiatry 41: 15–22.
Bunney WE, Bunney BG (2000). Molecular clock genes in man and lower animals: possible implications for circadian abnormalities in depression. Neuropsychopharmacology 22: 335–345.
Burt DB, Zembar MJ, Niederehe G (1995). Depression and memory impairment: a meta-analysis of the association, its pattern, and specificity. Psychol Bull 117: 285–305.
Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW et al (2002). Role of genotype in the cycle of violence in maltreated children. Science 297: 851–854.
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H et al (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301: 386–389.
Charney DS (1998). Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry 59: 11–14.
Charney DS, Barlow DH, Botteron KN, Cohen JD, Goldman D, Gur RC et al (2002). Neuroscience research agenda to guide development of a pathophysiologically based classification system. In: Kupfer DJ, First MB, Regier DA (eds). A Research Agenda for DSM-V. American Psychiatric Association: Washington, DC. pp 31–83.
Charney DS, Drevets WC (2002). The neurobiological basis of anxiety disorders. In: Davis KL, Charney DS, Coyle JT, Nemeroff CB (eds). Neuropsychopharmacology: The Fifth Generation of Progress. Lippincott Williams & Wilkins: Philadelphia, PA. pp 901–930.
Charney DS, Menkes DB, Heninger GR (1981). Receptor sensitivity and the mechanism of action of antidepressant treatment. Implications for the etiology and therapy of depression. Arch Gen Psychiatry 38: 1160–1180.
Chen AC, Shirayama Y, Shin KH, Neve RL, Duman RS (2001). Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biol Psychiatry 49: 753–762.
Claes S, Villafuerte S, Forsgren T, Sluijs S, Del-Favero J, Adolfsson R et al (2003). The corticotropin-releasing hormone binding protein is associated with major depression in a population from Northern Sweden. Biol Psychiatry 54: 867–872.
Comings DE, Wu S, Rostamkhani M, McGue M, Iacono WG, MacMurray JP (2002). Association of the muscarinic cholinergic 2 receptor (CHRM2) gene with major depression in women. Am J Med Genet 114: 527–529.
Coppen A, Eccleston EG, Peet M (1973). Total and free tryptophan concentration in the plasma of depressive patients. Lancet 2: 60–63.
Costa Jr PT, McCrae RR (1988). Personality in adulthood: a six-year longitudinal study of self-reports and spouse ratings on the NEO Personality Inventory. J Pers Soc Psychol 54: 853–863.
Costello EJ, Pine DS, Hammen C, March JS, Plotsky PM, Weissman MM et al (2002). Development and natural history of mood disorders. Biol Psychiatry 52: 529–542.
Damasio AR, Tranel D, Damasio H (1990). Individuals with sociopathic behavior caused by frontal damage fail to respond autonomically to social stimuli. Behav Brain Res 41: 81–94.
de Kloet RE (2003). Hormones, brain and stress. Endocr Regul 37: 51–68.
Delgado PL, Miller HL, Salomon RM, Licinio J, Krystal JH, Moreno FA et al (1999). Tryptophan-depletion challenge in depressed patients treated with desipramine or fluoxetine: implications for the role of serotonin in the mechanism of antidepressant action. Biol Psychiatry 46: 212–220.
Delgado PL, Price LH, Miller HL, Salomon RM, Aghajanian GK, Heninger GR et al (1994). Serotonin and the neurobiology of depression. Effects of tryptophan depletion in drug-free depressed patients. Arch Gen Psychiatry 51: 865–874.
Drevets WC (2000). Neuroimaging studies of mood disorders. Biol Psychiatry 48: 813–829.
Drevets WC (2001). Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol 11: 240–249.
Drevets WC, Frank E, Price JC, Kupfer DJ, Greer PJ, Mathis C (2000). Serotonin type-1A receptor imaging in depression. Nucl Med Biol 27: 499–507.
Drevets WC, Frank E, Price JC, Kupfer DJ, Holt D, Greer PJ et al (1999). PET imaging of serotonin 1A receptor binding in depression. Biol Psychiatry 46: 1375–1387.
Drevets WC, Price JL, Simpson Jr JR, Todd RD, Reich T, Vannier M et al (1997). Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386: 824–827.
Drevets WC, Raichle ME (1998). Reciprocal suppression of regional cerebral blood flow during emotional versus higher cognitive processes: implications for interactions between emotion and cognition. Cognition Emotion 12: 353–385.
Drevets WC, Sills R, Nugent AC, Bain E, Neumeister A, Price J et al (2004). Volumetric assessment of the amygdala in mood disorders using high resolution, 3T MRI. Biol Psychiatry 55: 182S.
Drevets WC, Videen TO, Price JL, Preskorn SH, Carmichael ST, Raichle ME (1992). A functional anatomical study of unipolar depression. J Neurosci 12: 3628–3641.
Dryman A, Eaton WW (1991). Affective symptoms associated with the onset of major depression in the community: findings from the US National Institute of Mental Health Epidemiologic Catchment Area Program. Acta Psychiatr Scand 84: 1–5.
Duncan Jr WC (1996). Circadian rhythms and the pharmacology of affective illness. Pharmacol Ther 71: 253–312.
Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN (2003). Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 60: 804–815.
Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE et al (2001). Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci USA 98: 6917–6922.
Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A et al (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112: 257–269.
Elliott R, Rubinsztein JS, Sahakian BJ, Dolan RJ (2002). The neural basis of mood-congruent processing biases in depression. Arch Gen Psychiatry 59: 597–604.
Fanous A, Gardner CO, Prescott CA, Cancro R, Kendler KS (2002). Neuroticism, major depression and gender: a population-based twin study. Psychol Med 32: 719–728.
Farmer A, Mahmood A, Redman K, Harris T, Sadler S, McGuffin P (2003). A sib-pair study of the Temperament and Character Inventory scales in major depression. Arch Gen Psychiatry 60: 490–496.
Fawcett J, Clark DC, Scheftner WA, Gibbons RD (1983). Assessing anhedonia in psychiatric patients. Arch Gen Psychiatry 40: 79–84.
Francis DD, Meaney MJ (1999). Maternal care and the development of stress responses. Curr Opin Neurobiol 9: 128–134.
Gass P, Kretz O, Wolfer DP, Berger S, Tronche F, Reichardt HM et al (2000). Genetic disruption of mineralocorticoid receptor leads to impaired neurogenesis and granule cell degeneration in the hippocampus of adult mice. EMBO Rep 1: 447–451.
Geiman TM, Robertson KD (2002). Chromatin remodeling, histone modifications, and DNA methylation—how does it all fit together? J Cell Biochem 87: 117–125.
Gesing A, Bilang-Bleuel A, Droste SK, Linthorst AC, Holsboer F, Reul JM (2001). Psychological stress increases hippocampal mineralocorticoid receptor levels: involvement of corticotropin-releasing hormone. J Neurosci 21: 4822–4829.
Giasson BI, Lee VM (2003). Are ubiquitination pathways central to Parkinson's disease? Cell 114: 1–8.
Giles DE, Jarrett RB, Rush AJ, Biggs MM, Roffwarg HP (1993). Prospective assessment of electroencephalographic sleep in remitted major depression. Psychiatry Res 46: 269–284.
Giles DE, Kupfer DJ, Rush AJ, Roffwarg HP (1998). Controlled comparison of electrophysiological sleep in families of probands with unipolar depression. Am J Psychiatry 155: 192–199.
Gillard ER, Dang DQ, Stanley BG (1993). Evidence that neuropeptide Y and dopamine in the perifornical hypothalamus interact antagonistically in the control of food intake. Brain Res 628: 128–136.
Glazier AM, Nadeau JH, Aitman TJ (2002). Finding genes that underlie complex traits. Science 298: 2345–2349.
Gold PW, Drevets WC, Charney DS (2002). New insights into the role of cortisol and the glucocorticoid receptor in severe depression. Biol Psychiatry 52: 381–385.
Gold PW, Loriaux DL, Roy A, Kling MA, Calabrese JR, Kellner CH et al (1986). Responses to corticotropin-releasing hormone in the hypercortisolism of depression and Cushing's disease. Pathophysiologic and diagnostic implications. N Engl J Med 314: 1329–1335.
Gottesman II, Gould TD (2003). The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 160: 636–645.
Gottesman II, Shields J (1973). Genetic theorizing and schizophrenia. Br J Psychiatry 122: 15–30.
Graves LA, Hellman K, Veasey S, Blendy JA, Pack AI, Abel T (2003). Genetic evidence for a role of CREB in sustained cortical arousal. J Neurophysiol 90: 1152–1159.
Griens AM, Jonker K, Spinhoven P, Blom MB (2002). The influence of depressive state features on trait measurement. J Affect Disord 70: 95–99.
Gross C, Zhuang X, Stark K, Ramboz S, Oosting R, Kirby L et al (2002). Serotonin1A receptor acts during development to establish normal anxiety-like behaviour in the adult. Nature 416: 396–400.
Hagerty BM, Williams RA, Liken M (1997). Prodromal symptoms of recurrent major depressive episodes: a qualitative analysis. Am J Orthopsychiatry 67: 308–314.
Hammen C, Marks T, Mayol A, deMayo R (1985). Depressive self-schemas, life stress, and vulnerability to depression. J Abnorm Psychol 94: 308–319.
Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D et al (2002). Serotonin transporter genetic variation and the response of the human amygdala. Science 297: 400–403.
Hariri AR, Weinberger DR (2003). Imaging genomics. Br Med Bull 65: 259–270.
Hasler G, Pine DS, Gamma A, Milos G, Ajdacic V, Eich D et al (2004). The association between psychopathology and being overweight. Psychol Med (in press).
Heim C, Nemeroff CB (2001). The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry 49: 1023–1039.
Heuser I, Yassouridis A, Holsboer F (1994). The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J Psychiatr Res 28: 341–356.
Hirayasu Y, Shenton ME, Salisbury DF, Kwon JS, Wible CG, Fischer IA et al (1999). Subgenual cingulate cortex volume in first-episode psychosis. Am J Psychiatry 156: 1091–1093.
Holsboer F, Lauer CJ, Schreiber W, Krieg JC (1995). Altered hypothalamic-pituitary-adrenocortical regulation in healthy subjects at high familial risk for affective disorders. Neuroendocrinology 62: 340–347.
Jang KL, Livesley WJ, Vernon PA (1996). Heritability of the big five personality dimensions and their facets: a twin study. J Pers 64: 577–591.
Karten YJ, Nair SM, van Essen L, Sibug R, Joels M (1999). Long-term exposure to high corticosterone levels attenuates serotonin responses in rat hippocampal CA1 neurons. Proc Natl Acad Sci USA 96: 13456–13461.
Keating M, Atkinson D, Dunn C, Timothy K, Vincent GM, Leppert M (1991). Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science 252: 704–706.
Keating MT, Sanguinetti MC (2001). Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104: 569–580.
Kegeles LS, Malone KM, Slifstein M, Ellis SP, Xanthopoulos E, Keilp JG et al (2003). Response of cortical metabolic deficits to serotonergic challenge in familial mood disorders. Am J Psychiatry 160: 76–82.
Kendell RE (1989). Clinical validity. Psychol Med 19: 45–55.
Kendler KS, Davis CG, Kessler RC (1997). The familial aggregation of common psychiatric and substance use disorders in the National Comorbidity Survey: a family history study. Br J Psychiatry 170: 541–548.
Kendler KS, Eaves LJ, Walters EE, Neale MC, Heath AC, Kessler RC (1996). The identification and validation of distinct depressive syndromes in a population-based sample of female twins. Arch Gen Psychiatry 53: 391–399.
Kendler KS, Gardner CO, Prescott CA (1999). Clinical characteristics of major depression that predict risk of depression in relatives. Arch Gen Psychiatry 56: 322–327.
Kendler KS, Thornton LM, Prescott CA (2001). Gender differences in the rates of exposure to stressful life events and sensitivity to their depressogenic effects. Am J Psychiatry 158: 587–593.
Klaassen T, Riedel WJ, Deutz NE, Van Praag HM (2002). Mood congruent memory bias induced by tryptophan depletion. Psychol Med 32: 167–172.
Klein DF (1974). Endogenomorphic depression. A conceptual and terminological revision. Arch Gen Psychiatry 31: 447–454.
Koed K, Linnet K (2000). Opposing changes in serotonin and norepinephrine transporter mRNA levels after serotonin depletion. Eur Neuropsychopharmacol 10: 501–509.
Koper JW, Stolk RP, de Lange P, Huizenga NA, Molijn GJ, Pols HA et al (1997). Lack of association between five polymorphisms in the human glucocorticoid receptor gene and glucocorticoid resistance. Hum Genet 99: 663–668.
Koschack J, Hoschel K, Irle E (2003). Differential impairments of facial affect priming in subjects with acute or partially remitted major depressive episodes. J Nerv Ment Dis 191: 175–181.
Kuroda Y, Watanabe Y, Albeck DS, Hastings NB, McEwen BS (1994). Effects of adrenalectomy and type I or type II glucocorticoid receptor activation on 5-HT1A and 5-HT2 receptor binding and 5-HT transporter mRNA expression in rat brain. Brain Res 648: 157–161.
Lambert G, Johansson M, Agren H, Friberg P (2000). Reduced brain norepinephrine and dopamine release in treatment-refractory depressive illness: evidence in support of the catecholamine hypothesis of mood disorders. Arch Gen Psychiatry 57: 787–793.
Landro NI, Stiles TC, Sletvold H (2001). Neuropsychological function in nonpsychotic unipolar major depression. Neuropsychiatry Neuropsychol Behav Neurol 14: 233–240.
Lemonde S, Turecki G, Bakish D, Du L, Hrdina PD, Bown CD et al (2003). Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J Neurosci 23: 8788–8799.
Linkowski P, Van Onderbergen A, Kerkhofs M, Bosson D, Mendlewicz J, Van Cauter E (1993). Twin study of the 24-h cortisol profile: evidence for genetic control of the human circadian clock. Am J Physiol 264: E173–E181.
Liotti M, Mayberg HS, McGinnis S, Brannan SL, Jerabek P (2002). Unmasking disease-specific cerebral blood flow abnormalities: mood challenge in patients with remitted unipolar depression. Am J Psychiatry 159: 1830–1840.
Loas G, Boyer P, Legrand A (1999). Anhedonia in the deficit syndrome of schizophrenia. Psychopathology 32: 207–219.
Lopez JF, Chalmers DT, Little KY, Watson SJ (1998). A.E. Bennett Research Award. Regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol Psychiatry 43: 547–573.
Luciano M, Wright MJ, Smith GA, Geffen GM, Geffen LB, Martin NG (2003). Genetic covariance between processing speed and IQ In: Plomin R, DeFries JC, Craig IW, McGuffin P (eds). Behavioral Genetics in the Postgenomic Era. American Psychological Association: Washington, DC. pp 163–181.
Lyons DM, Yang C, Sawyer-Glover AM, Moseley ME, Schatzberg AF (2001). Early life stress and inherited variation in monkey hippocampal volumes. Arch Gen Psychiatry 58: 1145–1151.
MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe RT et al (2003). Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci USA 100: 1387–1392.
MacQueen GM, Galway TM, Hay J, Young LT, Joffe RT (2002). Recollection memory deficits in patients with major depressive disorder predicted by past depressions but not current mood state or treatment status. Psychol Med 32: 251–258.
Malhotra AK, Kestler LJ, Mazzanti C, Bates JA, Goldberg T, Goldman D (2002). A functional polymorphism in the COMT gene and performance on a test of prefrontal cognition. Am J Psychiatry 159: 652–654.
Manji HK, Drevets WC, Charney DS (2001). The cellular neurobiology of depression. Nat Med 7: 541–547.
Manji HK, Duman RS (2001). Impairments of neuroplasticity and cellular resilience in severe mood disorders: implications for the development of novel therapeutics. Psychopharmacol Bull 35: 5–49.
Manji HK, Gottesman II, Gould TD (2003a). Signal transduction and genes-to-behaviors pathways in psychiatric diseases. Sci STKE 2003: pe49.
Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, N AG et al (2003b). Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 53: 707–742.
Mattay VS, Goldberg TE, Fera F, Hariri AR, Tessitore A, Egan MF et al (2003). Catechol O-methyltransferase val158-met genotype and individual variation in the brain response to amphetamine. Proc Natl Acad Sci USA 100: 6186–6191.
Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA et al (1999). Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry 156: 675–682.
McEwen BS (1999). Stress and hippocampal plasticity. Annu Rev Neurosci 22: 105–122.
McMahon SD, Grant KE, Compas BE, Thurm AE, Ey S (2003). Stress and psychopathology in children and adolescents: is there evidence of specificity? J Child Psychol Psychiatry 44: 107–133.
Meguid MM, Fetissov SO, Varma M, Sato T, Zhang L, Laviano A et al (2000). Hypothalamic dopamine and serotonin in the regulation of food intake. Nutrition 16: 843–857.
Mendels J, Frazer A (1974). Brain biogenic amine depletion and mood. Arch Gen Psychiatry 30: 447–451.
Merikangas KR, Zhang H, Avenevoli S, Acharyya S, Neuenschwander M, Angst J (2003). Longitudinal trajectories of depression and anxiety in a prospective community study: the Zurich Cohort Study. Arch Gen Psychiatry 60: 993–1000.
Miller HL, Delgado PL, Salomon RM, Berman R, Krystal JH, Heninger GR et al (1996a). Clinical and biochemical effects of catecholamine depletion on antidepressant-induced remission of depression. Arch Gen Psychiatry 53: 117–128.
Miller HL, Delgado PL, Salomon RM, Heninger GR, Charney DS (1996b). Effects of alpha-methyl-para-tyrosine (AMPT) in drug-free depressed patients. Neuropsychopharmacology 14: 151–157.
Mitchell P, Hadzi-Pavlovic D, Parker G, Hickie I, Wilhelm K, Brodaty H et al (1996). Depressive psychomotor disturbance, cortisol, and dexamethasone. Biol Psychiatry 40: 941–950.
Mizoguchi K, Ishige A, Aburada M, Tabira T (2003). Chronic stress attenuates glucocorticoid negative feedback: involvement of the prefrontal cortex and hippocampus. Neuroscience 119: 887–897.
Modell S, Huber J, Holsboer F, Lauer CJ (2003). The Munich Vulnerability Study on Affective Disorders: risk factors for unipolarity versus bipolarity. J Affect Disord 74: 173–184.
Modell S, Lauer CJ, Schreiber W, Huber J, Krieg JC, Holsboer F (1998). Hormonal response pattern in the combined DEX–CRH test is stable over time in subjects at high familial risk for affective disorders. Neuropsychopharmacology 18: 253–262.
Modell S, Yassouridis A, Huber J, Holsboer F (1997). Corticosteroid receptor function is decreased in depressed patients. Neuroendocrinology 65: 216–222.
Moreno FA, Rowe DC, Kaiser B, Chase D, Michaels T, Gelernter J et al (2002). Association between a serotonin transporter promoter region polymorphism and mood response during tryptophan depletion. Mol Psychiatry 7: 213–216.
Muller M, Holsboer F, Keck ME (2002). Genetic modification of corticosteroid receptor signalling: novel insights into pathophysiology and treatment strategies of human affective disorders. Neuropeptides 36: 117–131.
Murck H, Nickel T, Kunzel H, Antonijevic IA, Schill J, Zobel A et al (2003). State markers of depression in sleep EEG: dependency on drug and gender in patients treated with tianeptine or paroxetine. Neuropsychopharmacology 28: 348–358.
Murphy FC, Sahakian BJ, Rubinsztein JS, Michael A, Rogers RD, Robbins TW et al (1999). Emotional bias and inhibitory control processes in mania and depression. Psychol Med 29: 1307–1321.
Murphy FC, Smith KA, Cowen PJ, Robbins TW, Sahakian BJ (2002). The effects of tryptophan depletion on cognitive and affective processing in healthy volunteers. Psychopharmacology (Berl) 163: 42–53.
Nathan PJ, Stough C (2001). Inspection time: a neuropsychophysiological test for measuring the functional integrity of the cholinergic system. Med Hypotheses 57: 759–760.
Nelson JC, Charney DS (1981). The symptoms of major depressive illness. Am J Psychiatry 138: 1–13.
Nemeroff CB (1996). The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry 1: 336–342.
Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M (1988). Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry 45: 577–579.
Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K et al (1984). Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science 226: 1342–1344.
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002). Neurobiology of depression. Neuron 34: 13–25.
Nestler EJ, Barrot M, Self DW (2001). DeltaFosB: a sustained molecular switch for addiction. Proc Natl Acad Sci USA 98: 11042–11046.
Neumeister A, Bain E, Nugent AC, Carson RE, Bonne O, Luckenbaugh DA et al (2004). Reduced serotonin type 1A receptor binding in panic disorder. J Neurosci 24: 589–591.
Neumeister A, Konstantinidis A, Stastny J, Schwarz MJ, Vitouch O, Willeit M et al (2002). Association between serotonin transporter gene promoter polymorphism (5HTTLPR) and behavioral responses to tryptophan depletion in healthy women with and without family history of depression. Arch Gen Psychiatry 59: 613–620.
Neumeister A, Turner EH, Matthews JR, Postolache TT, Barnett RL, Rauh M et al (1998). Effects of tryptophan depletion vs catecholamine depletion in patients with seasonal affective disorder in remission with light therapy. Arch Gen Psychiatry 55: 524–530.
Nofzinger EA, Nichols TE, Meltzer CC, Price J, Steppe DA, Miewald JM et al (1999). Changes in forebrain function from waking to REM sleep in depression: preliminary analyses of [18F]FDG PET studies. Psychiatry Res 91: 59–78.
Odagaki Y, Garcia-Sevilla JA, Huguelet P, La Harpe R, Koyama T, Guimon J (2001). Cyclic AMP-mediated signaling components are upregulated in the prefrontal cortex of depressed suicide victims. Brain Res 898: 224–231.
Öngür D, Drevets WC, Price JL (1998). Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA 95: 13290–13295.
Oquendo MA, Barrera A, Ellis SP, Li S, Burke AK, Grunebaum M et al (2004). Instability of symptoms in recurrent major depression: a prospective study. Am J Psychiatry 161: 255–261.
Parker G (2000). Classifying depression: should paradigms lost be regained? Am J Psychiatry 157: 1195–1203.
Petronis A (2001). Human morbid genetics revisited: relevance of epigenetics. Trends Genet 17: 142–146.
Petronis A, Gottesman II, Kan P, Kennedy JL, Basile VS, Paterson AD et al (2003). Monozygotic twins exhibit numerous epigenetic differences: clues to twin discordance? Schizophr Bull 29: 169–178.
Pizzagalli DA, Oakes TR, Fox AS, Chung MK, Larson CL, Abercrombie HC et al (2004). Functional but not structural subgenual prefrontal cortex abnormalities in melancholia. Mol Psychiatry 9: 325.
Raadsheer FC, Hoogendijk WJ, Stam FC, Tilders FJ, Swaab DF (1994). Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology 60: 436–444.
Rao U, Dahl RE, Ryan ND, Birmaher B, Williamson DE, Rao R et al (2002). Heterogeneity in EEG sleep findings in adolescent depression: unipolar versus bipolar clinical course. J Affect Disord 70: 273–280.
Redei EE, Ahmadiyeh N, Baum AE, Sasso DA, Slone JL, Solberg LC et al (2001). Novel animal models of affective disorders. Semin Clin Neuropsychiatry 6: 43–67.
Riedel WJ, Klaassen T, Schmitt JA (2002). Tryptophan, mood, and cognitive function. Brain Behav Immun 16: 581–589.
Riemann D, Berger M, Voderholzer U (2001). Sleep and depression—results from psychobiological studies: an overview. Biol Psychol 57: 67–103.
Riemann D, Hohagen F, Krieger S, Gann H, Muller WE, Olbrich R et al (1994). Cholinergic REM induction test: muscarinic supersensitivity underlies polysomnographic findings in both depression and schizophrenia. J Psychiatr Res 28: 195–210.
Rogers MA, Bradshaw JL, Phillips JG, Vaddadi K, Presnel I, Mileshkin C (2000). Parkinsonian motor characteristics in unipolar major depression. J Clin Exp Neuropsychol 22: 232–244.
Rogers RD, Tunbridge EM, Bhagwagar Z, Drevets WC, Sahakian BJ, Carter CS (2003). Tryptophan depletion alters the decision-making of healthy volunteers through altered processing of reward cues. Neuropsychopharmacology 28: 153–162.
Roiser JP, Rubinsztein JS, Sahakian BJ (2003). Cognition in depression. In: Spilberg H (eds). Psychiatry. Medicine Publishing Company: Abingdon.
Salomon RM, Miller HL, Krystal JH, Heninger GR, Charney DS (1997). Lack of behavioral effects of monoamine depletion in healthy subjects. Biol Psychiatry 41: 58–64.
Semont A, Fache M, Ouafik L, Hery M, Faudon M, Hery F (1999). Effect of serotonin inhibition on glucocorticoid and mineralocorticoid expression in various brain structures. Neuroendocrinology 69: 121–128.
Sen S, Nesse RM, Stoltenberg SF, Li S, Gleiberman L, Chakravarti A et al (2003). A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology 28: 397–401.
Sheline YI (2003). Neuroimaging studies of mood disorder effects on the brain. Biol Psychiatry 54: 338–352.
Sheline YI, Mintun MA (2002). Structural and functional imaging of affectie disorders In: Nemeroff CB (eds). Neuropsychopharmacology: The Fifth Generation of Progress. Lippincott Williams & Wilkins: Philadelphia, PA. pp 1065–1080.
Sheline YI, Sanghavi M, Mintun MA, Gado MH (1999). Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci 19: 5034–5043.
Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS (2002). Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 22: 3251–3261.
Siegle GJ, Konecky RO, Thase ME, Carter CS (2003). Relationships between amygdala volume and activity during emotional information processing tasks in depressed and never-depressed individuals: an fMRI investigation. Ann NY Acad Sci 985: 481–484.
Sobin C, Sackeim HA (1997). Psychomotor symptoms of depression. Am J Psychiatry 154: 4–17.
Stroud LR, Salovey P, Epel ES (2002). Sex differences in stress responses: social rejection versus achievement stress. Biol Psychiatry 52: 318–327.
Stunkard AJ, Fernstrom MH, Price A, Frank E, Kupfer DJ (1990). Direction of weight change in recurrent depression. Consistency across episodes. Arch Gen Psychiatry 47: 857–860.
Sullivan PF, Neale MC, Kendler KS (2000). Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 157: 1552–1562.
Sullivan RM, Gratton A (1999). Lateralized effects of medial prefrontal cortex lesions on neuroendocrine and autonomic stress responses in rats. J Neurosci 19: 2834–2840.
Taheri S, Mignot E (2002). The genetics of sleep disorders. Lancet Neurol 1: 242–250.
Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK et al (1998). Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet 19: 162–166.
Toczek MT, Carson RE, Lang L, Ma Y, Spanaki MV, Der MG et al (2003). PET imaging of 5-HT1A receptor binding in patients with temporal lobe epilepsy. Neurology 60: 749–756.
Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM et al (2001). An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 291: 1040–1043.
Tremblay LK, Naranjo CA, Cardenas L, Herrmann N, Busto UE (2002). Probing brain reward system function in major depressive disorder: altered response to dextroamphetamine. Arch Gen Psychiatry 59: 409–416.
Trichard C, Martinot JL, Alagille M, Masure MC, Hardy P, Ginestet D et al (1995). Time course of prefrontal lobe dysfunction in severely depressed in-patients: a longitudinal neuropsychological study. Psychol Med 25: 79–85.
Tsourtos G, Thompson JC, Stough C (2002). Evidence of an early information processing speed deficit in unipolar major depression. Psychol Med 32: 259–265.
Tsuang MT, Faraone SV, Lyons MJ (1993). Identification of the phenotype in psychiatric genetics. Eur Arch Psychiatry Clin Neurosci 243: 131–142.
Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W et al (2001). Brain dopamine and obesity. Lancet 357: 354–357.
Watkins PC, Vache K, Verney SP, Muller S, Mathews A (1996). Unconscious mood-congruent memory bias in depression. J Abnorm Psychol 105: 34–41.
Young EA (1998). Sex differences and the HPA axis: implications for psychiatric disease. J Gend Specif Med 1: 21–27.
Young EA, Lopez JF, Murphy-Weinberg V, Watson SJ, Akil H (2003). Mineralocorticoid receptor function in major depression. Arch Gen Psychiatry 60: 24–28.
Zetterstrom TS, Pei Q, Madhav TR, Coppell AL, Lewis L, Grahame-Smith DG (1999). Manipulations of brain 5-HT levels affect gene expression for BDNF in rat brain. Neuropharmacology 38: 1063–1073.
Zobel AW, Nickel T, Kunzel HE, Ackl N, Sonntag A, Ising M et al (2000). Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res 34: 171–181.
Zobel AW, Yassouridis A, Frieboes RM, Holsboer F (1999). Prediction of medium-term outcome by cortisol response to the combined dexamethasone–CRH test in patients with remitted depression. Am J Psychiatry 156: 949–951.
Zubenko GS, Hughes III HB, Maher BS, Stiffler JS, Zubenko WN, Marazita ML (2002). Genetic linkage of region containing the CREB1 gene to depressive disorders in women from families with recurrent, early-onset, major depression. Am J Med Genet 114: 980–987.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hasler, G., Drevets, W., Manji, H. et al. Discovering Endophenotypes for Major Depression. Neuropsychopharmacol 29, 1765–1781 (2004). https://doi.org/10.1038/sj.npp.1300506
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300506
Keywords
This article is cited by
-
Methods for quantifying the heterogeneity of psychopathology
BMC Psychiatry (2023)
-
Altered hippocampal subfield volumes in major depressive disorder with and without anhedonia
BMC Psychiatry (2023)
-
Familial risk for depression is associated with reduced P300 and late positive potential to affective stimuli and prolonged cardiac deceleration to unpleasant stimuli
Scientific Reports (2023)
-
Sleep disturbance as a transdiagnostic marker of psychiatric risk in children with neurodevelopmental risk genetic conditions
Translational Psychiatry (2023)
-
Revisiting the theoretical and methodological foundations of depression measurement
Nature Reviews Psychology (2022)
