
Abstract
This study examined the effects of topiramate (TPM) on glucocorticoid receptors (GRs) in mononuclear leukocytes of nine men and four women with chronic and recurring post-traumatic stress disorder (PTSD) and a group of comparison subjects (nine men, four women). A measure of 60 ml of blood was withdrawn by venipuncture at 0800 and mononuclear leukocytes were isolated. The cells were incubated with a series of concentrations of dexamethasone (DEX) without or with 50 μmol/l of TPM to evaluate the effects of DEX to inhibit lysozyme activity and the effect of TPM on it. ANCOVA compared the IC50 for lysozyme inhibition under conditions of DEX only and TPM+DEX. TPM affected lysozyme IC50 in the direction of increasing the sensitivity of the receptor in the sample as a whole. This effect was more pronounced in the mononuclear leukocytes from participants in the PTSD group, particularly in cells from subjects whose pretreatment lysozyme IC50 was relatively higher (eg, reflecting decreased glucococorticoid receptor responsiveness), compared to the rest of the sample. In conclusion, further investigation of the actions of TPM on GR and other neuroendocrine systems may prove useful in understanding some of the other established clinical effects of this agent.
Similar content being viewed by others
INTRODUCTION
Although the mechanism of action of topiramate (TPM) has not yet been fully characterized, laboratory and clinical studies have provided substantial evidence for the ‘antikindling’ and neuroprotective effects of TPM. In laboratory studies, TPM blocked the effects of kainite-induced excitatory conductance in cultured rat hippocampal neurons (Gibbs et al, 2000), and reduced the effects of hippocampal neuronal damage in gerbils caused by ischemia (Lee et al, 2000). These and other preclinical findings have provided a putative mechanism for the effects of TPM as an anticonvulsant (Jahromi et al, 2000; Shank et al, 2000), and are consistent with some clinical reports of the antiepileptic effects and mood-stabilizing effects of this compound (Chengappa et al, 1999; Jette et al, 2002; Privitera et al, 2003).
TPM has been recently suggested in the treatment of post-traumatic stress disorder (PTSD) (Berlant and van Kammen, 2000), a disorder that has been associated with, among other things, hippocampal degeneration (eg Schuff et al, 2001). Although the mechanisms involved in PTSD-related hippocampal degeneration are not well understood, many investigators have speculated that cortisol alterations, and, in particular, changes in the glucocorticoid receptor (GR), may play a role (Sapolsky, 2000; Yehuda, 2001). The cascade of events that endangers neurons once GRs are activated is complex, but appears to involve the effects of cortisol, which, at high levels, inhibits the reuptake mechanism that removes glutamate from the synapse and, accordingly, increases the activation of postsynaptic N-methyl-D-aspartic acid (NMDA) (Sapolsky, 1994). The activation of NMDA receptors results in a series of events (eg, increased concentration of intracellular calcium, proteolysis, potentiation of oxidative damage) that ultimately endanger the cell (Landfield and Eldridge, 1994).
Alterations in the responsiveness of hippocampal GR may have implications for changes in hippocampal neuronal survival induced by NMDA receptor activation. Although TPM potentiates GABAergic neurotransmission (Deutsch et al, 2002) and antagonizes the excitotoxic actions of glutamate at the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA)/kainate (KA) classes of glutamate-gated channels, this effect could potentially be amplified if TPM also exerted direct effects on GR. Interestingly, a paradoxical neuroendocrine effect of TPM has also been observed in that this drug was found to reduce corticosterone levels and mRNA expression for CRH, but was also found to reduce, rather than increase, mRNA expression for Type 2 GRs in the hypothalamic tissue of rats fed a high-fat diet (York, 2000).
In view of this possibility, and the potential importance of glucocorticoid-related alterations in both neurodegenerative and psychiatric disorders that might be ameliorated by neuroprotective agents, we examined the effect of TPM on glucocorticoid responsiveness in mononuclear leukocytes, as expressed by the lysozyme IC50. One function of glucocorticoids is to inhibit the expression of the lysozyme gene; thus, exposing mononuclear leukocytes to DEX enhances the inhibition of lysozyme activity, and provides a direct measure of glucocorticoid sensitivity (Panarelli et al, 1994). In the current study, mononuclear leukocytes were isolated, and treated with DEX and with or without 50 μmol/l of TPM to determine whether TPM would alter the lysozyme IC50.
Since little is currently known about the endocrine effects of TPM, it was of interest to examine the effects of this agent in healthy persons without medical or psychiatric disorder, and also in a group of patients with lifetime PTSD and other comorbid mood and anxiety disorders, whom we suspected would show alterations in the sensitivity of GR.
METHODS
Subjects
A total of 26 subjects were studied. Recruitment was through advertisements placed in local newspapers or on bulletin boards around the Mount Sinai Hospital and the Bronx Veteran Affairs Medical Center (VAMC) requesting volunteers for research examining hormonal responses following extremely stressful life events. All procedures were approved by the Institutional Review Boards at the Mount Sinai School of Medicine and the Bronx Veterans Affairs, and all subjects provided written informed consent prior to participation. Healthy controls were free from current or lifetime Axis I disorder. Subjects with past, but not current, substance dependence were included if they were in sustained full remission. Subjects in the PTSD group were excluded if they had an Axis I disorder other than PTSD, unless this disorder was a comorbid mood or anxiety disorder. Also excluded were participants with major medical, endocrinological, or neurological disorder likely to interfere with HPA axis function. Subjects were not withdrawn from medications to participate in the study. However, because we wished to eliminate confounds associated with the effects of other medications, we excluded subjects who were taking any type of medication for medical or psychiatric illness.
Clinical Assessments
Information about traumatic life events was obtained using the Trauma History Questionnaire (THQ) (Green, 1996), and were reviewed with the subject in order to ascertain whether they met the diagnostic criteria, ‘Criterion A,’ for a traumatic event (ie, involved actual or perceived threat of serious harm, and provoked a response of intense fear, helplessness, or horror) (American Psychiatric Association, 1994). Subjects who endorsed a Criterion A event were evaluated with the Clinician Administered PTSD Scale (CAPS) (Blake et al, 1995), using the most disturbing experience as the basis of the CAPS interview. Other Axis I diagnoses were made using the Structured Clinical Interview for DSM-IV (Spitzer et al, 1995). Diagnostic interviews were conducted by a trained psychologist or psychiatrist with established inter-rater reliability, and diagnoses were confirmed by consensus conference.
Laboratory Methods
Plasma and lymphocytes preparation
A measure of 60 ml blood was drawn in tubes containing EDTA at 0800. Cells were separated from platelet-rich plasma by centrifuging at 300g for 15 min at room temperature. After adding the same volume of Hanks balanced salt solution (HBS) as plasma had been aspirated, the lymphocytes were isolated by Ficoll-Paque gradient method (Ficoll-Paque PLUS, Amershan Pharmacia Biotech, Piscataway, NJ) using ACCUSPIN Tube (Sigma, St Louis, MO). After washing two times in ice-cold HBS, 6–8-ml of cold HBS was added into the pellet tube. A measure of 100 μl of the suspension was counted by a hemocytometer; 1–2 ml of the suspension, usually containing 0.5–2 × 107 cells, was pelleted by centrifuging and stored at −70°C for GR binding assay. The rest of the cell suspension was repelleted and resuspended in cold RPMI-1640 medium supplemented with 10% (V/V) fetal bovine sera, 50 U/ml penicillin and 50 μg/ml streptomycin (Gibco, Life Technologies Ltd., Grand Island, NY). The concentration of cells was 3.5–4.0 × 105 cells/150 μl used for the lysozyme inhibition test. The platelet-rich plasma was centrifuged at 2700g for 15 min at 4°C. The platelet-poor plasma was collected for cortisol assay.
GR binding assay
Receptor binding was measured with the ligand 3H-dexamethasone (3H-DEX, Amershan Pharmacia Biotech, Piscataway, NJ) according to our published method (Yehuda et al, 1995). The interassay and intra-assay coefficients of variation were 5.0 and 8.5%, respectively.
Plasma cortisol assay
Plasma cortisol was determined by radioimmunoassay (RIA) using the ‘GammaCoat Cortisol 125I RIA Kit’ (DiaSorin, Stillwater, MN). The interassay and intra-assay coefficients of variation were 3.0 and 7.7%, respectively.
Inhibition of lysozyme synthesis by DEX and the effect of TPM
We modified the method for assessment of inhibition of lysozyme synthesis by DEX described by Panarelli et al (1994). In brief, 20 μl of different concentrations of DEX in HBS were added into 96 flat-bottomed wells of a microplate to give a final concentration of 0.5–100 nmol/l. HBS (20 μl) was added to a control well. Supplemented RPMI-1640 (50 μl) without or with TPM (Johnson & Johnson Pharmaceutical Research & Development, LLC, Raritan, NJ) was added to give a final concentration of TPM of 50 μmol/l. A measure of 150 μl of cell suspension as prepared above was placed in each well. Each sample was run in triplicate. After mixing for 1 min, the plate was incubated for 72 h in 5% CO2/air at 37°C in a humidified chamber. After centrifuging the plate, a 120 μl aliquot of supernatant was removed and pooled from each of the triplicate wells. The supernatant was stored at −70°C until lysozyme assay. Using this preparation we have previously found that lysozyme IC50 correlates with the cortisol response to DEX in healthy (eg without medical or psychiatric disorder) volunteers (Yehuda, 2003).
Lysozyme activity in supernatant was measured by the turbidimetric method. Micrococcus lysodeikticus (Sigma, St Louis, MO) was used as a substrate, which was prepared in 0.1 mol/l phosphate buffer (pH 6.3). A measure of 20 μl of supernatant was mixed with 150 μl of M. lysodeikticus in a microplate. Each sample was assayed in triplicate. The plate was incubated at 37°C for 30 min and read optical density (OD) at 450 nm by a Bio-Rad plate-reader (Bio-Rad Laboratories, Richmond, CA) or was preincubated for 7–10 min, and then kinetically read by a VERSAmax microplate reader (Molecular Devices, Sunnyvale, CA) at 37°C for 20 min. The interassay and intra-assay coefficients of variation for the measurement of lysozyme activity were 4.5 and 9.2%, respectively.
Calculations for activity of lysozyme
The relative percent of lysozyme activity (RAL %) was used for creating a DEX dose–inhibiting curve. The calculation for RAL % in DEX only was as follows:

For cells pretreated with TPM (DEX+TPM) the calculation for RAL% was as follows:

The DEX concentration that would give 50% inhibition of lysozyme activity (IC50 DEX) was determined from the DEX dose–response curve by GraFit software (Sigma, St Louis, MO). The interassay and intra-assay coefficients of variation were 5.0 and 8.5%, respectively.
Statistical analysis
Group differences in IC50 with and without TPM, cortisol levels and GR number were examined using analysis of variance or covariance (ANOVA or ANCOVA). For the primary analyses comparing the IC50 response to TPM between the two groups, repeated measures ANCOVA was used, with the within-subjects factor being Condition (ie IC50 with and without TPM). In all ANCOVAs, the homogeneity of the covariance coefficients was verified. The Group by Condition interaction was evaluated to examine group differences in the relative change in IC50 with TPM. Regression analyses were used to confirm the importance of significant covariates with respect to their contribution to group differences in IC50.
Initial analyses were carried out to identify potential confounds in the data. Age, gender, weight, height, body mass index (BMI), depression, anxiety, and substance abuse comorbidity were tested individually for association with the biological outcome measures (IC50 with and without TPM, baseline cortisol level, and GR binding). Based on correlational analyses, age, height, and weight were not significantly associated with these measures. BMI was significantly associated with IC50 in the DEX+TPM condition (r=−0.43, n=26, p=0.026) and was therefore used as a covariate. Based on one-way ANOVA, there were no effects of gender, lifetime depression, anxiety disorder, or substance abuse on any of the biological variables.
When the same covariates were considered within the repeated measures ANCOVA, lifetime anxiety disorder and lifetime MDD both showed significant interactions with Condition and, therefore, were also used as covariates in those analyses.
RESULTS
Demographic and clinical characteristics of the sample are reported in Table 1.
TPM treatment had the effect of increasing the inhibitory effects of DEX on lysozyme IC50 (mean (SD) without TPM: 6.7 μmol/l (2.3); with TPM: 5.7 μmol/l (2.5)); however, in the absence of controlling for any potentially confounding variable, this effect only reached a trend level of significance (F(1,25)=3.6, p=0.069). After controlling for the presence of any current or lifetime psychiatric disorder (eg PTSD, depression, or other mood disorder, any anxiety disorders, past substance abuse, or dependence), this effect reached statistical significance (F(1,21)=6.23, p=0.021). All covariates except substance abuse were significant.
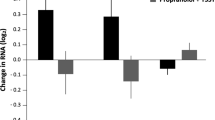
Figure 1 shows the inhibition of lysozyme activity to DEX and the effect of TPM to influence this effect in human peripheral mononuclear leukocytes in the two groups, PTSD vs nonpsychiatric comparison subjects. The graph demonstrates a subtle effect of TPM to ‘shift’ the curve towards increasing the inhibitory effects of DEX. When the repeated measures ANCOVA was performed using the presence or absence of PTSD as a main effect, there remained a significant main effect of TPM (F(1,22)=35.56, p<0.0005), controlling for lifetime history of depression and anxiety disorder. This effect persisted when BMI was also added as a covariate (F(1,21)=9.39, p=0.006), and reflected a lower IC50 when cells were incubated with DEX+TPM compared to when they were incubated with DEX alone (adjusted mean (SE): 6.71 μmol/l (0.41) vs 5.70 μmol/l (0.44)). Repeated measures ANCOVA also demonstrated a significant Group by Condition interaction [F(1,21)=34.81, p<0.0005), but no significant main effect of Group (F(1,21)=0.24, p=0.63). BMI was not associated with IC50 in this analysis, but there was a significant BMI by Condition interaction (F(1,21)=5.23, p=0.03). The presence of lifetime MDD (F(1,21)=24.43, p<0.0005) and anxiety disorder (F(1,21)=22.22, p<0.0005) both showed a significant interaction with Condition. In the absence of these covariates, this interaction was no longer significant.

Inhibition of lysozyme activity by DEX and the effect of TPM in human peripheral lymphocytes from 13 control (a) and 13 PTSD (b) subjects. In all, 3.5−4.0 × 105 cells were incubated with various levels of DEX without or with TPM (50 μmol/l) in RPMI-1640 medium for 72 h. See Methods for details.
The Group by Condition interaction reflects that those with lifetime PTSD demonstrated a greater reduction overall in IC50 with TPM than the comparison group. IC50 started out higher in subjects with PTSD, but decreased when cells were incubated with TPM (adjusted mean (SE) of IC50 in the PTSD group in cells incubated with DEX only was 8.84 μmol/l (0.85) and 4.27 μmol/l (0.91) in cells incubated with DEX+TPM). In contrast, the adjusted means for the comparison group demonstrate an elevation in IC50 in the DEX+TPM condition (adjusted mean 4.59 μmol/l (0.85) vs 7.13 μmol/l (0.91) in the DEX only and DEX+TPM conditions, respectively).
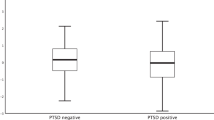
Figure 2 shows the individual data points from all subjects to illustrate that the mean trend is slightly misleading in that, in subjects with lifetime PTSD, the response to TPM was quite variable. Given that the presence or absence of comorbid mood and anxiety disorders was found to impact the data so substantially, we felt it necessary to perform a post hoc examination, beginning with visual inspection of the raw data, to determine the possibility of subgroup effects. Figure 2 clearly demonstrates that subjects who started out with higher IC50 values in the DEX-only condition tended to show lower values when cells were treated with TPM. In subjects who started out with lower IC50 values treatment with TPM tended to increase the IC50. The mean (SD) value of the absolute difference in IC50 was 2.8 (2.5) in the PTSD group and 1.1 (1.4) in comparison subjects.

Inhibition of lysozyme activity by dexamethasone and by dexamethasone+topiramate in peripheral lymphocytes for each of the comparison and PTSD subjects.
Analyses of baseline biological measures show that there were no significant group differences in 0800 cortisol level (F(1,22)=0.70, p=0.41) or GR number (F(1,22)=0.11, p=0.75) controlling for BMI. In the PTSD group, as compared to the comparison group, the adjusted mean cortisol (μg/dl) level was 9% lower (11.7 (0.91) vs 12.4 (0.91)) and the adjusted mean number of GR/cell was 11% greater (2870 (600) vs 2520 (600)). These differences were not significant. There was a significant partial correlation between cortisol and re-experiencing symptoms (r=−0.506, df=18, p=0.023), but no relationships were observed between symptom severity and IC50. There were no significant correlations among the major biologic variables (IC50 with and without TPM, cortisol, GR).
DISCUSSION
The current findings demonstrate that TPM can affect the activity of the GR. The main overall effect of this drug was to decrease IC50. This effect was particularly pronounced in the group with lifetime PTSD and, in particular, in the mononuclear leukocytes of those with PTSD whose pretreatment lysozyme IC50 was high. A high IC50 is consistent with diminished receptor sensitivity (Panarelli et al, 1994). Diminished GR sensitivity has been associated with MDD (for a review see Pariante and Miller, 2001). That TPM might increase the sensitivity of GR is consistent with observed effects of this compound in mood disorders (Chengappa et al, 1999; Grunze et al, 2001; Carpenter et al, 2002). On the other hand, in some subjects, primarily those with PTSD whose pretreatment lysozyme IC50 values were relatively low, TPM had the effect of reducing the sensitivity of the receptor, similar to what was observed in rats who demonstrated decreased mRNA expression following treatment with TPM. A low IC50 reflects enhanced receptor sensitivity (Panarelli et al, 1994). Evidence for enhanced receptor sensitivity has been observed in PTSD (Yehuda et al, 1995; Yehuda, 2002). That TPM can reduce the sensitivity of GR is theoretically consistent with therapeutic effectiveness in PTSD, in so far as GR sensitivity plays a role in the pathophysiology of this disorder. In subjects without psychiatric disorder, the effect of TPM was negligible.
One possible mechanism for the effect of TPM is in reducing the efficiency of the membrane transporters, such as P-glycoprotein, that transports DEX out of the cell (eg Schinkel et al, 1995; Meijer et al, 1998) conceivably resulting in an apparent reduction in IC50 in the assay. This effect would be consistent with observations with respect to the activity of some antidepressants (Pariante and Miller, 2001). However, one of the observations in this study is that there was no uniform effect of TPM in the group with PTSD. This was an unanticipated finding as we had hypothesized, on the basis of our prior studies demonstrating enhanced receptor responsiveness in PTSD (Yehuda et al, 1995; Yehuda, 2002) that the effect of TPM would be to increase IC50, and had not anticipated the wide variation in the effects of this drug in the PTSD group. The participants in the PTSD group were carefully selected on the basis of having been exposed to extremely traumatic events and having chronic and recurrent PTSD symptoms for several years or decades. They were somewhat different from other groups of PTSD subjects we have studied in the past in being more heterogeneous with respect to age, gender, trauma type, and diagnostic comorbidity. These variables might account for the variability of the HPA axis alterations observed in this sample. Indeed, in this relatively small and heterogeneous PTSD sample, we did not observe significantly lower cortisol levels or a significantly greater number of GR on mononuclear leukocytes, although the mean values were in the direction of effects observed in prior studies. Yet, all participants were symptomatic on multiple domains of PTSD, depressive and anxiety-related symptoms. That TPM can have differential effects on the sensitivity of GR in patients within the broad diagnostic category of PTSD based on whether IC50 was high or low suggests that TPM may act in a restorative capacity, rather than to either systematically reduce or increase lysozyme IC50. These actions should be investigated further.
The dose of TPM used in this study, 50 μmol/l, was at the low end of the recommended therapeutic range, and only a single dose was used. This, too, might account for the variable and/or relatively modest effects of TPM. Other studies of the mechanism of action of TPM in cell preparations found effects at doses beginning at 100 μmol/l. The effect of TPM in decreasing mRNA of hypothalamic Type II GR in rats occurred after a daily regimen of 40 mg/kg/day for 80 days. Certainly, the effects of chronic TPM may differ from the effects of a single, relatively low dose administered to live cells. Furthermore, the effects of TPM on GR in blood cells may be vastly different from its effects on GR in both endocrine or brain tissue. That a measurable signal could be detected, however, suggests that the current methodology might be a useful probe in drug development and, more specifically, in evaluating the effects of a compound on GR-mediated activity. Future studies might compare effects observed in the current study with endocrine effects observed when TPM is ingested either acutely or chronically, or may use this methodology to test the effect of other agents on GR activity in vitro.
There is good reason to suspect that the effects described are due to the effects of TPM on the GR-mediated-activity, and the use of DEX decreases the likelihood that the effects are mediated through the mineralocorticoid receptor. However, in view of the fact that the complex glucocorticoid signaling cascade is subject to modulation at so many molecular levels, starting with extra- and intracellular hormone availability, and ending with modification at the DNA level with steroid receptor coactivators/repressors, it is important to not attribute changes in DEX modulation of lysozyme production as owing solely to changes in GR sensitivity, but also, more generally, as glucocorticoid sensitivity.
If TPM does affect GR, this might explain the effectiveness of this agent in a wide range of clinical and metabolic syndromes, including eating disorders and obesity, in which there are alterations of the HPA axis. Alternatively, GRs might be affected as a consequence of other properties of TPM, such as its ability to block binding to glutamate receptors in the brain, enhance the inhibitory activity of brain γ-aminobutyric acid (GABA), or block sodium channels (De Lorenzo et al, 2000). The current study suggests that an examination of the effects of TPM on GC may prove useful in understanding some of the other established effects of this agent in neuroprotection, and their putative neuroendocrine links.
References
American Psychiatric Association (1994). Diagnostic and Statistical Manual IV. American Psychiatric Press: Wahington, DC.
Berlant J, van Kammen DP (2000). Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry 63: 15–20.
Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS (1995). The development of a Clinician-Administered PTSD Scale. J Trauma Stress 8: 75–90.
Carpenter LL, Leon Z, Yasmin S, Price LH (2002). Do obese depressed patients respond to topiramate? A retrospective chart review. J Affect Disord 69: 251–255.
Chengappa KN, Rathore D, Levine J, Atzert R, Solai L, Parepally H 1999. Topiramate as add-on treatment for patients with bipolar mania. Bipolar Disord 1: 42–53.
De Lorenzo RJ, Sombati S, Coulter DA. (2000). Effects of topiramate on sustained repetitive firing and spontaneous recurrent seizure discharges in cultured hippocampal neurons. Epilepsia 41: S40–S44.
Deutsch SI, Rosse RB, Billingslea EN, Ellack AS, Mastropaolo J (2002). Topiramates antagonizes MK-801 in an animal model of schizophrenia. Eur J Pharmacol 449: 121–125.
Gibbs III JW, Sombati S, DeLorenzo RJ, Coulter DA (2000). Cellular actions of topiramate: blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia 41: S10–S16.
Green BL (1996). In: Stamm BH (ed). Trauma History Questionnaire in Measurement of Stress, Trauma, and Adaptation. Sidran: Lutherville, MD.
Grunze HC, Normann C, Langosch J, Schaefer M, Amann B, Sterr A (2001). Antimanic efficacy of topiramate in 11 patients in an open trial with an on-off-on design. J Clin Psychiatry 62: 464–468.
Jahromi SS, Pelletier MR, McDonald PJ, Khosravani H, Carlen PL 2000. Antiepileptic efficacy of topiramate: assessment in two in vitro seizure models. Brain 87: 20–28.
Jette NJ, Marson AG, Hutton JL 2002. Topiramate add-on for drug-resistant partial epilepsy. Cochrane Database System Rev, CD001417.
Landfield PW, Eldridge JC 1994. The glucocorticoid hypothesis of age-related hippocampal neurodegeneration: role of dysregulated intraneuronal calcium. Ann NY Acad Sci 74: 308–321.
Lee SR, Kim SP, Kim JE (2000). Protective effect of topiramate against hippocampal neuronal damage after global ischemia in the gerbils. Neurosci Lett 281: 183–186.
Meijer OC, deLange ECM, Breimer DD, Boer AG, deKloet ER (1998). Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdr1A P-glycoprotein knockout mice. Endocrinology 139: 1789–1998.
Panarelli M, Holloway CD, Mulatero P, Fraser R, Kenyon J (1994). Inhibition of lysozyme synthesis by dexamethasone in human mononuclear leukocytes: an index of glucocorticoid sensitivity. J Clin Endocrinol Metab 78: 872–877.
Pariante CM, Miller AH (2001). Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry 49: 391–404.
Privitera MD, Brodie MJ, Mattson RH, Chadwick DW, Neto W, Wang S (2003). EPMN 105 Study Group. Acta Neurol Scand 107: 165–175.
Sapolsky RM (1994). The physiological relevance of glucocorticoid endangerment of the hippocampus. Ann NY Acad Sci 746: 294–304.
Sapolsky RM. (2000). Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 57: 925–935.
Schinkel AH, Wagenaar E, van Deemter L, Mol CAAM, Borst P (1995). Absence of the mdr1a P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest 96: 1698–1705.
Schuff N, Neylan TC, Lenoci MA, Du AT, Weiss DS, Marmar CR (2001). Decreased hippocampal N-acetylaspartate in the absence of atrophy in posttraumatic stress disorder. Biol Psychiatry 50: 952–959.
Shank RP, Gardocki JF, Streeter AF, Maryanoff BE (2000). An overview of the preclinical aspects of topiramate: pharmacology, pharmacokinetics, and mechanism of action. Epilepsia 41: S3–S9.
Spitzer RL, Williams JBW, Gibbon M (1995). Structured Clinical Interview for DSM-IV (SCID). New York State Psychiatric Institute, Biometrics Research: New York.
Yehuda R (2001). Are glucocorticoids responsible for putative hippocampal damage in PTSD? How and when to decide. Hippocampus 11: 85–89.
Yehuda R (2002). Current status of cortisol findings in post-traumatic stress disorder. Psychiatr Clin North Am 25: 341–368.
Yehuda R, Boisoneau D, Lowy MT, Giller Jr EL (1995). Dose–response changes in plasma cortisol and lymphocyte glucocorticoid receptors following dexamethasone administration in combat veterans with and without posttraumatic stress disorder. Arch Gen Psychiatry 52: 583–593.
Yehuda R, Halligan SL, Grossman R, Golier JA, Wong C (2002). The cortisol and glucocorticoid receptor response to low dose dexamethasone administration in aging combat veterans and holocaust survivors with and without posttraumatic stress disorder. Biological Psychiatry 52: 393–403.
Yehuda R, Makotkine I, Ling SG, Singh B, Yang RK (2003). Relationship between dexamethasone-inhibited lysozyme activity in peripheral mononuclear leukocytes and the cortisol and glucocorticoid receptor response to dexamethasone. J Psychiatric Res 34: 471–477.
York DA (2000). Effect of topiramate on body weight and body composition of Osborne-Mendel rats fed a high fat diet: alterations in hormones, neuropeptide and uncoupling protein mRNAs. Nutrition 16: 967–975.
Acknowledgements
This study was supported by a contract from Johnson & Johnson Pharmaceutical Research & Development, LLC. We thank Daniel van Rammen and Richard Shank for their thoughtful comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yehuda, R., Yang, RK., Golier, J. et al. Effect of Topiramate on Glucocorticoid Receptor Mediated Action. Neuropsychopharmacol 29, 433–439 (2004). https://doi.org/10.1038/sj.npp.1300331
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300331
Keywords
This article is cited by
-
The Antidepressant Desipramine Requires the ABCB1 (Mdr1)-Type p-Glycoprotein to Upregulate the Glucocorticoid Receptor in Mice
Neuropsychopharmacology (2007)
-
Effect of Sertraline on Glucocorticoid Sensitivity of Mononuclear Leukocytes in Post-Traumatic Stress Disorder
Neuropsychopharmacology (2006)
