
Abstract
Currently, most basic and clinical research on depression is focused on either central serotonergic, noradrenergic, or dopaminergic neurotransmission as affected by various etiological and predisposing factors. Recent evidence suggests that there is another system that consists of a subset of brain α1B-adrenoceptors innervated primarily by brain epinephrine (EPI) that potentially modulates the above three monoamine systems in parallel and plays a critical role in depression. The present review covers the evidence for this system and includes findings that brain α1-adrenoceptors are instrumental in behavioral activation, are located near the major monoamine cell groups or target areas, receive EPI as their neurotransmitter, are impaired or inhibited in depressed patients or after stress in animal models, and are restored by a number of antidepressants. This ‘EPI-α1 system’ may therefore represent a new target system for this disorder.
Similar content being viewed by others


INTRODUCTION
Depressive illness is currently believed to result from environmental (stress) and genetically induced alterations in neurotransmission in one or more of the central monoaminergic pathways (noradrenergic, serotonergic, and dopaminergic) mediated by a complex web of factors including corticotropin-releasing factor, hypothalamic–pituitary–adrenal hormones, and cytokines (Anisman et al, 2002; Nemeroff, 2002; Nestler et al, 2002). With regard to monoaminergic function, most recent basic and clinical research has been focused on the serotonergic system owing to the success of serotonin-selective antidepressants, while the role of the noradrenergic function has been relatively neglected. However, recent work by our group and others, considered in the light of past research, has provided compelling new evidence linking one of the brain's adrenoceptors (α1) and the ‘minor’ brain neurotransmitter, epinephrine (EPI), to this disorder. This evidence suggests that there is a putative EPI-innervated α1-receptor system that is critical for the regulation of active behavior and functions by causing a parallel excitation of the three major monoaminergic systems. This new system appears to be impaired during stress and depression and thus may represent a new target for this disorder. The evidence is reviewed here.
ROLE OF α1-ADRENOCEPTORS IN BEHAVIORAL ACTIVITY
The new evidence largely concerns a clarification of the role of α1-adrenoceptors in behavioral activity. One of the core behavioral symptoms of depression is a profound loss of interest in previously pleasurable, nonessential activities. Particularly impaired are those activities that are mediated by positive motivation and entail a fair degree of sustained effort such as hobbies and avocations, sports, socialization, entertainment, ‘going out’, etc.
From a neurobiological viewpoint, a reduction in positively motivated effortful behavioral activity would most likely be caused by a reduction of central dopaminergic neurotransmission in basal ganglia circuits. In agreement, a number of studies have shown that there is a reduced level of DA and its metabolites in the CSF of depressed patients (Brown and Gershon, 1993) and also a reduced release and metabolism of DA in the basal ganglia of animals subjected to stressors that induce behavioral depression (Cabib and Puglisi-Allegra, 1996). This reduction, however, does not appear to be caused by a direct impairment of the dopaminergic neurons or postsynaptic receptors themselves (Weiss et al, 1996; Cabib et al, 1998; Ossowska et al, 2001; Herman et al, 1984; Zebrowska-Lupina et al, 1988), but may occur instead in an afferent system that modulates dopaminergic function. One such afferent system that is particularly sensitive to stress and has been implicated in depressive illness is the α1-adrenergic (Lipinski et al, 1987; Stone and Quartermain, 1999).
α1-Adrenoceptors are G-protein coupled receptors that are widely distributed in the CNS and periphery and that are involved in a wide variety of behavioral, autonomic, and neuroendocrine functions. These include motor activity (see Stone et al, 1999 for references), attention and vigilance (Aston-Jones et al, 1994), fear and anxiety (Arnsten et al, 1999; Fujimaki et al, 2000; Yang et al, 1990), reinforcement (Woolverton, 1987), sensory gating (Bakshi and Geyer, 1999), learning (Ferry et al, 1999; MacDonald and Sirvio, 1999), sexual behavior (Chu and Etgen, 1999), and regulation of blood pressure (Hieble et al, 1999) and of both the hypothalamic–pituitary–adrenal (Feuvrier et al, 1998) and hypothalamic–pituitary–gonadal systems (Dubocovich et al, 1990).
It is currently thought that the α1-receptors involved in motor activity exert this function in large part via modulatory effects on dopaminergic neuronal firing rate (Berretta et al, 2000; Grenhoff et al, 1993, 1995), presynaptic release of DA (Gioanni et al, 1998; Mathé et al, 1996; Auclair et al, 2002), and/or potentiation of postsynaptic dopaminergic signaling (Eshel et al, 1990). However, DA-independent effects may also be involved since many α1-receptors are located on motorneurons downstream of dopaminergic neurons (Hou et al, 2002) and also on or near serotonergic and noradrenergic neurons (Morin et al, 1996).
In previous years, it had been difficult to study these α1-receptors because prazosin, the prototypical α1-antagonist used by most investigators, is poorly soluble in saline and cannot be given centrally in high enough doses to block the brain's population of these receptors. To address this problem, our group began to study another α1-antagonist, terazosin, which has the same receptor selectivity as prazosin but is 20 times more soluble.
The initial experiment was a dose–response analysis in mice of the effect of intraventricular (ivt.) terazosin on behavioral activity stimulated by change to a novel cage (Stone et al, 1999). This ‘cage-change’ test produced sustained exploration in mice for 1–2 h with minimal anxiety, since the novel cage was the same type as that which the animal was housed in. Terazosin produced a striking dose-dependent complete inhibition of all movement, leaving the animals cataleptic at the highest dose (31.6 nmol/mouse). This effect was selective to the α1-receptor blocker, since similar experiments with an α2- (RX821002), β1- (betaxolol), or β2-antagonist (ICI118551) produced weaker or no effects. The terazosin-treated mice were not significantly sedated since they showed normal righting reflexes and could support themselves for 30 s on a taut horizontal wire; by contrast, akinetic chlordiazepoxide-treated mice fell off within 10 s. The animals showed a slowed respiratory rate and hypothermia but had only a slight reduction in blood pressure (5 mmHg).
To verify that the immobility was the result of α1-receptor blockade rather than some nonspecific toxic effect of the drug, the agonist, phenylephrine (PE), was coinjected with the terazosin to determine if it would reverse the inactivity, which it did. Furthermore, we showed that terazosin, at the highest dose, retained its selectivity for α1 vs DAergic or α2-receptors, as it did not protect D1-receptors from in vivo alkylation by N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) (but did protect α1-receptors), did not displace a D2-specific ligand from striatal D2-receptors up to 10−4 M, and did not reduce the ex vivo binding of an α2-specific ligand, but did abolish that of an α1 ligand in the cerebral cortex.
To determine how closely the loss of active behavior paralleled the blockade of brain α1-receptors, a range of doses of terazosin was injected ivt. and the resulting behavioral inactivity scores of the animals were correlated with measures of the ex vivo binding of [3H]prazosin to α1-receptors in their cerebral cortices, which were obtained immediately after the behavioral test (Stone et al, 2001c). A very high and significant correlation was obtained (r=0.95). This experiment was next repeated with i.p. prazosin instead of ivt. terazosin. The former drug had to be given at very high doses (16–32 mg/kg) in order to pass the blood–brain barrier in these animals. (The low doses of prazosin generally used in behavioral research (0.5–2.0 mg/kg) did not significantly reduce ex vivo α1-binding in the cerebral cortex of our mice (male Swiss Webster) in support of similar findings by others for the mouse (Lindroos et al, 1984) and dog (Taylor et al, 1977).) A similar and near-perfect correlation was obtained again between loss of α1-binding sites and loss of active behavior, which made it virtually certain that these receptors controlled behavioral activity in this species. These close relations between behavior and α1-receptor binding were in agreement with earlier studies of α1-binding in the spinal cord and startle behavior (Astrachan et al, 1983a, 1983b).
We next attempted to determine which subtype of α1-receptor mediated the motor activation. The mammalian brain is known to possess the genes and express the mRNA of three α1-receptors, 1A, 1B, and 1D. The proteins for only two of these (α1A and α1B) have so far been found in brain tissue (Garcia-Sainz et al, 1999; Schwinn, 2000; Yang et al, 1998). No highly specific antagonists of these subtypes were available that could be used to unambiguously identify the responsible receptor. Therefore a series of α1-antagonists having a range of affinities for the subtypes was injected ivt and their affinities to inhibit active behavior in the novel cage test (inhibitory dose 50%) was correlated with their known affinities to bind to the cloned receptors (Ki values). A high correlation would identify the receptor in question. Six antagonists were used: terazosin, benoxathian, phentolamine, 5-methylurapidil, BMY7378, and WB4101. Behavioral ID50 values were obtained from 9–11 point dose–response curves for each antagonist against activity in the novel cage test. Binding affinities were obtained from the literature by computing the average of all published values (3–11 studies) for these antagonists at cloned α1A-, α1B-, and α1D-receptors. The results showed a high and significant correlation for α1B (0.89), with none for either the α1A- (0.27) or α1D-receptors (0.13). This finding agreed with a previous study that showed that agonists of α1B-receptors, but not of α1A or α1D were effective in the reversal of cataplexy in narcoleptic dogs (Nishino et al, 1993).
These results, however, appeared to be in partial disagreement with findings from genetically altered mouse strains. An α1B-receptor deficient mouse strain had been generated (Cavalli et al, 1997) and had been reported to show ‘normal’ activity in a novel chamber or open field (Spreng et al, 2001); however, a second group did report a significant reduction in the open field activity with this strain (Knauber and Muller, 2000). In order to determine if there was compensation for the loss of the α1B-receptor in the mutant mice, we challenged them with large doses of the stimulant, modafinil. The latter drug appears to stimulate motor activity in part by activation of central α1-receptors (Duteil et al, 1990; Simon et al, 1995; Stone et al, 2002a). The mutants were found to be less than half as responsive as their wild-type conspecifics to the highest modafinil dose (40 mg/kg) (Stone et al, 2002a). Drouin et al (2002) had performed a similar experiment using three other stimulants also believed to act in part via α1-receptors, amphetamine, cocaine, and morphine, and had found the α1 mutants to be much less active than wild-type controls in response to each given i.p. In a further preliminary experiment, we found that ivt. terazosin can completely inhibit novel cage activity in the α1B mutant mice (Stone EA, Lin Y, and Quartermain D, unpublished findings), suggesting that the compensation is mediated by another α1-receptor.
Blendy and colleagues have recently generated an α1D-deficient strain and have reported that these mice show reduced rearing responses in a novel cage and reduced nocturnal wheel running in the home cage (Saldalge et al, 2002). Our inability to detect the role of the α1D-receptor may be explainable by the fact that no one has yet succeeded in demonstrating any α1D-binding in the brain of either the rat (Yang et al, 1997), mouse (Yang et al, 1998), or rabbit (Piao et al, 2000) using the above antagonists as displacing agents. Therefore the α1D-receptor responsible for behavioral activation must have some unique property such as a potential location in the peripheral nervous system which in some way communicates with the CNS.
The above experiments therefore established that central α1-adrenoceptors are essential for behavioral activation in response to either novelty or psychostimulant drugs. The primary subtype involved in the mouse brain is the α1B-adrenoceptor but a second central α1-receptor, as yet unidentified, can partially compensate for its loss during development.
BRAIN LOCALIZATION
A fundamental question regarding the motoric α1-receptors concerns the brain sites that they operate in. α1-Receptors are present in a broad range of structures including most motor, sensory, autonomic, and neuroendocrine-related areas (Acosta-Martinez et al, 1999; Day et al, 1997; Jones et al, 1985; Volgin et al, 2001; Williams and Morilak, 1997; Zilles et al, 1993). They are present at low densities in basal ganglia structures (corpus striatum, nucleus accumbens, ventral tegmental area, substantia nigra), at higher densities in prefrontal and motor cortices, hypothalamus, brainstem motor nuclei, cerebellum, and spinal motorneurons, and at very high densities in the olfactory bulb, thalamus, locus coeruleus (LC), dorsal raphe (DR), piriform cortex, and amygdala. There is a species difference with regard to the hippocampus, which has a very low concentration in rodents, but in primates, including humans, has a very high concentration of these receptors primarily in the dentate gyrus (Palacios et al, 1987).
To investigate the localization, we tested the ability of microinjections of terazosin in discrete regions of the mouse brain to induce immobility in a 10 min exposure to a novel cage (Lin et al, 2002; Lin and Stone, 2001). At present we have tested 23 brain areas of which four have given definite positive responses (ie inactivity) and two potentially positive responses. The definite areas are the dorsal pons in or near the LC, the DR, the nucleus accumbens, and the cerebellum lobules lying dorsal to the fourth ventricle. Positive responses have also been obtained from the IV ventricle which suggests diffusion to nearby periventricular sites of either or both the dorsal pons and cerebellum. The potential areas are the medullary C2 area and medial preoptic area in which partial responses have been encountered.
It should be noted that two of the above definite sites, the dorsal pons and DR, are known to possess very high densities of α1-receptors (Chamba et al, 1991; Jones et al, 1985; Zilles et al, 1993), whereas the cerebellum has been reported by some (Acosta-Martinez et al, 1999; Jones et al, 1985; Zilles et al, 1993), but not others (Palacios et al, 1987), to have a moderately high density. We have confirmed that there is a moderately high density of α1-receptors in the cerebellum of the mouse strain used in the above experiments (male Swiss Webster) (Stone EA, Ahsan R, and Lin Y, unpublished results). As noted above, the nucleus accumbens also possesses α1-receptors but at a lower density (Jones et al, 1985).
To control for nonspecific effects, we have shown further that the terazosin-induced immobility at each site can be reversed completely by coinfusion of the full α1/α2-agonist, 6-fluoronorepinephrine. A selective full α2-agonist, dexmedetomidine, was ineffective in reversing the terazosin immobility. The selective α1-agonist, PE, has been less reliable in reversing this immobility, probably owing to the fact that it is only a partial agonist at rodent brain α1-receptors mediating phosphatidylinositol hydrolysis or potentiation of cAMP responses (Johnson and Minneman, 1986).
Owing to the small size of the mouse brain and the possibility of diffusion of terazosin to noninjected sites, the above conclusions remain tentative and will need to be confirmed by other methods and in larger animals. Nevertheless, the nucleus accumbens findings agree with previous work by Cools (1991), who has shown that stimulation of α1-receptors in this area elicits motor activity in the rat. Others have found evidence for motoric α1-receptors in the rat prefrontal cortex (Blanc et al, 1994; Gioanni et al, 1998; Trovero et al, 1992) and hippocampus (Plaznik et al, 1984). However, we have not found terazosin to induce immobility in either of these structures of the mouse brain, but this may be the result of the use of stimulant drugs or different behavioral tests, time periods, and species.
Surprisingly, we found that the terazosin produced the opposite effect, that is, an enhancement of motor activity, when injected into the dorsal hippocampus or lateral septum. This suggests that α1-receptors in the latter two regions suppress rather than excite behavioral activity. A similar effect had been reported by Kelsey (1976) for the rat septum. As will be discussed in the next section, the latter findings may be relevant to the role of α1-receptors in vigilance and anxiety.
In preliminary work, we have examined the ability of α1-agonists injected alone locally into these regions to induce behavioral activation in animals inactive in their home cages during the light period. So far we have found that 6FNE injected into the dorsal pons elicits sustained motor and exploratory behavior in the home cage (Stone EA, Lin Y, and Quartermain D, unpublished findings). The partial agonist PE has been less reliable in this regard.
The finding of positive motor sites in the DR, dorsal pons, and nucleus accumbens suggests that α1-receptors regulate motor activity via effects on three major monoaminergic systems of the brain: the serotonergic, noradrenergic, and dopaminergic. Stimulation of α1-receptors can produce excitation of neurons in the DR (Baraban and Aghajanian, 1980; Menkes et al, 1981; Ohliger-Frerking et al, 2002; Reinhard Jr et al, 1983; Rouqier et al, 1994), the LC (Nakamura et al, 1988; Williams and Marshall, 1987), and the pontine reticular formation (Stevens et al, 1994) and, as discussed above, can facilitate the release of DA in the nucleus accumbens. In addition, neurons in the vermis cortex of the cerebellum are known to send efferent fibers to the LC and ventral tegmental area (Snider et al, 1976) and to the DR (Chan-Palay, 1977), which regulate the activity of these monoaminergic nuclei (Cano et al, 1980; Snider and Snider, 1979). The above brain localization of the motoric α1-receptors is consistent with the hypothesis that the α1-system coordinates and controls behavioral activation.
EPI AS AN ENDOGENOUS NEUROTRANSMITTER AT α1-RECEPTORS
A second fundamental question regarding this system concerns the identity of its endogenous catecholamine neurotransmitter. It has been widely assumed that it is NE, since the latter is the chief endogenous α1-agonist in the brain and since early studies showed that ivt. NE injections elicited motor activity (Geyer et al, 1972; Segal et al, 1974) via an α-receptor (Kleinrok and Zebrowska-Lupina, 1971; Zebrowska-Lupina et al, 1977), while electrolytic lesions of the LC impaired motor activity (Donaldson et al, 1976; Kostowski et al, 1978). However, a role for NE was not supported by many subsequent lesion studies, showing that the destruction of central noradrenergic pathways by the more selective neurotoxin, 6-hydroxydopamine (6-OHDA), did not induce hypoactivity in rats (McNaughton and Mason, 1980) and that a virtually complete elimination of this catecholamine from the brain produced by multiple-site central 6-OHDA lesions, failed to affect the nocturnal activity of rats (Murrough et al, 2000). However, it should be noted that compensatory changes are known to occur after these lesions and that the lesion of the dorsal noradrenergic bundle by 6-OHDA can potentiate the catalepsy induced by morphine (but not by neuroleptics) (Mason et al, 1978). These findings suggest that NE has a limited role in neurotransmission at motoric α1-receptors.
On the other hand, there is accumulating evidence of the fact that EPI is an important transmitter for these receptors. Although the tissue concentration of EPI is only a few percent of that of NE, the extracellular concentration of EPI in the rat hypothalamus measured by microdialysis (Routledge and Marsden, 1987a) or push-pull cannula (Phillipu et al, 1979) is equal to that of NE. EPI has traditionally been thought to be the endogenous ligand for brain postsynaptic α2-adrenoceptors, based on the finding that α2-receptors are upregulated in the brainstems of rats genetically low in brain EPI (BUF strain) (Vantini et al, 1984) or in animals treated chronically with inhibitors of phenylethanolamine-N-methyl transferase (PNMT) (U'Prichard et al, 1988). However, both rats of the latter strain and animals treated with PNMT inhibitors also show an equal upregulation of the density of brainstem α1-adrenoceptors (presumably postsynaptic to the source of EPI) and, in addition, an enhanced motor response to ivt. catecholamine injection (Segal et al, 1975), suggesting that brain α1- as well as α2-adrenoceptors are innervated by EPI. This agrees with the finding that EPI has the highest efficacy of all tested catecholamines at α1-receptors coupled to phosphatidylinositol hydrolysis or potentiation of cAMP responses in rat brain slices (Johnson and Minneman, 1986). Furthermore, pharmacological inhibition of PNMT, which causes a depletion of extracellular EPI but not of NE (Routledge and Marsden, 1987a), has been found to produce marked behavioral inactivity (Katz et al, 1978) and to abolish appetitive hypothalamic self-stimulation in rats and mice (Katz and Carroll, 1978), while stressors that release EPI, peripheral administration of EPI (Chopde et al, 1995; Yntema and Korf, 1987), and glucocorticoids that induce PNMT (Chopde et al, 1995) have all been found to reverse the catalepsy induced by neuroleptics probably via α1-receptor stimulation. Ivt. EPI administration has also been shown to markedly enhance hypothalamic self-stimulation in rats (Hasegawa, 1975).
Although the above studies showed that inhibition of PNMT produces behavioral inactivity and a concomitant depletion of extracellular EPI, they did not test whether the behavioral change was the result of the neurochemical one. To test this, we therefore determined if ivt. injection of EPI would selectively reverse the behavioral inactivity in PNMT-inhibited mice (Stone et al, in press). Mice given one of two PNMT inhibitors, 1,2-dichloro-α-methylbenzylamine (DCMB) or LY134046 received, after 30 min, an ivt. injection of EPI (1–100 nmol), and were screened for gross movement in a novel cage test for the following hour. In support of the hypothesis, exogenous EPI was found to dose-dependently reverse the PNMT-inhibitor-induced inactivity starting at about 3 nmol/mouse and reaching a maximal effect (60% reversal) at 10–30 nmol. It was also found that a higher dose of ivt. EPI (100 nmol) could reverse the behavioral inactivity of untreated animals in their home cages during the light period (10.00–12.00 h). The home cage during the light period is, of course, a low-stimulation environment for mice during the trough of their circadian activity cycle and is likely to produce a low release of brain EPI (Roth et al, 1982; Sauter et al, 1980). The reason why higher EPI doses were required to activate the home-caged compared to the PNMT-inhibited mice is most likely due to the fact that depletion of the transmitter sensitizes (ie reverses the ongoing desensitization of) the receptors (Katz et al, 1978; Watson et al, 2002).
Subsequent experiments with selective adrenoceptor antagonists infused ivt. with EPI showed that the above stimulatory effects of this amine in both the PNMT-inhibited and home-caged mice were due to activation of α1-receptors. Furthermore, it was found that, in addition to EPI, three other α1-agonists, NE, 6FNE, and PE also reversed the PNMT-inhibitor-induced inactivity and that the rank order of their maximal behavioral responses, EPI>6FNE>PE, matched the rank order of their maximal phosphatidylinositol hydrolysis responses in vitro in cells transfected with α1B-receptors (NE was not tested).
The fact that three other agonists also reversed the PNMT-inhibitor-induced inactivity, however, appeared to argue against the uniqueness of EPI as a transmitter for the above α1-receptors. This doubt was further reinforced by the additional finding that a noncatecholamine, 5-HT, was also capable of reversing DCMB inactivity. However, in support of EPI's role, we showed that its ability to reverse the DCMB-induced inactivity was the greatest of this group of agonists and also that EPI was the only agonist of the four that was capable of stimulating behavioral activity in the home-caged mice during the light period.
The above evidence therefore supports the hypothesis that EPI is an important endogenous catecholamine neurotransmitter at brain α1-receptors involved in motor activity. The neurophysiological significance of this finding may be elucidated by a comparison of the neuroanatomic distributions of motoric α1-adrenoceptors and EPI-containing neurons. As discussed above, motor-activating α1-receptors have been detected in the dorsal pons in or near the LC, DR, cerebellum, and nucleus accumbens with the first three regions also showing high or moderately high densities of α1-receptor binding sites. Also, as discussed above, the activation of α1-receptors in these brain regions is known to enhance neuronal firing rates and/or the release or postsynaptic action of their monoamine neurotransmitters (Rouqier et al, 1994; Stevens et al, 1994; Williams and Marshall, 1987). PNMT-containing nerve endings have been shown to innervate the LC and DR (Hokfelt et al, 1984), while both EPI and its metabolite, metanephrine, have been found to be markedly elevated in the accumbens during stimulant-induced hyperactivity (Espejo and Miñano, 2001).
These findings led us to propose that there is a central EPI-innervated α1-adrenergic system in the foregoing brain regions that activates motor behavior by producing either a parallel stimulation of three central monoaminergic systems, the noradrenergic, serotonergic, and dopaminergic, and/or an enhancement of their postsynaptic signaling (Figure 1). Serotonergic stimulation may predominate in this effect, since 5HT given without an α1-agonist was found in the above study to be capable of reversing the PNMT-inhibitor-induced inactivity (but not the home-cage light-phase-induced inactivity), whereas DA was not. Dopaminergic stimulation of active behavior may require the coactivation of postsynaptic α1- and DA-receptors to elicit the powerful synergy known to exist between the two on motor activity (Anden et al, 1973; Eshel et al, 1990). Noradrenergic activation may play a contributory role in behavioral activation or may be necessary to maintain arousal necessary for the latter to occur. As noted above, it cannot be decided from the present and previous findings whether NE itself contributes to the innervation of motoric α1-receptors in these regions and therefore this possibility remains open.

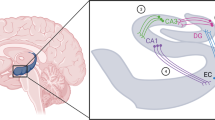
Cartoon showing relations between EPI innervation of α1-adrenoceptors in DR, dorsal pons–LC, nucleus accumbens (ACC), and cerebellum (CER). EPI is believed to either excite output neurons in these structures or facilitate the release of DA in the ACC via its actions on local α1-receptors. Also shown are efferent pathways from CER to the ventral tegmental area (VTA), DR, and LC. ? Indicates uncertainty that EPI is a neurotransmitter in CER which has a very low EPI tissue level.
As it currently stands, however, there are several problems with the foregoing hypothesis. First, the identity and properties of the neurons supplying EPI to the above brain regions is not yet clearly established. Second, excitation of EPI-containing neurons in the C1 area of the medulla has been found to inhibit rather than excite neuronal activity in the LC (Aston-Jones et al, 1992). Third, in the PNMT-inhibitor-treated mice ivt. EPI failed to restore rearing responses, a major component of exploratory behavior in rodents. Fourth, although EPI abolished immobility induced by PNMT-inhibition, it actually had a biphasic action reducing the latter behavior in doses up to about 10 nmol and then reinstating it at 30–100 nmol. Animals given the high doses showed alternating bouts of activity and immobility, and fifth, blockade of β1-adrenoceptors enhanced the reversal effect of EPI in the PNMT-inhibited mice and restored their rearing responses.
With respect to the identity of the EPI-supplying neurons, there appear to be at least two types of neurons that serve this function in the brain (see reviews by Mefford, 1988, 1987 and Fuller, 1982). The first, which is similar to classical monoaminergic neurons, contains all the catecholamine-synthesizing enzymes, including PNMT, and is capable of storing small but measurable amounts of EPI. These neurons predominate in the C1, C2, and C3 regions of the medulla and send axons to a variety of brainstem and forebrain structures including the LC, DR and, nucleus accumbens (Hokfelt et al, 1984). The second type of neuron is capable of synthesizing NE but does not contain PNMT. The latter enzyme is present in adjacent brain neurons and glia, which apparently synthesize EPI from released NE and transport it back to the NE-containing neurons, which store and utilize it as a cotransmitter of NE. This neuronal type predominates in the hypothalamus. It is presently not clear what the proportions are of these two neuronal types in the brain regions containing the motoric α1-receptors. It is also not established whether either or both of these EPI neurons possesses high-affinity uptake for EPI and to what extent they are sensitive to various uptake blocking drugs or to destruction by 6-OHDA or DSP4 which require uptake for their neurotoxicity (Fuller, 1982; Routledge and Marsden, 1987b). Blockade of monoamine oxidase A leads to a greater proportional rise in EPI compared to NE, however, the only pharmacological method presently available for selectively manipulating CNS EPI release independent of NE release is the inhibition of PNMT.
The finding that activation of EPI-containing neurons in the C1 area depresses LC unit activity appears to argue against a role of EPI in the activation of the LC-noradrenergic system. However, it should be noted that this finding was made in anesthetized rats and has not yet been shown in unanesthetized animals. Furthermore, there does not appear to be a tonic inhibition of LC neurons by EPI as blockade of PNMT with DCMB does not increase LC unit firing rate in rats (Engberg et al, 1981). Finally, the ability to elicit an α1-receptor-stimulated excitatory response from these neurons may also depend on the age of the animals and their hormonal status since LC excitation is readily observed in young animals (Williams and Marshall, 1987), and the expression of both α1B-receptors and PNMT are highly sensitive to induction by circulating levels of corticosterone (Sakaue and Hoffman, 1991; Stone and Kramer, 2001a; Wurtman, 2002).
With regard to the inability of EPI to restore rearing responses in PNMT-inhibited animals, this may have been due in part to the possibility that the exogenous catecholamine does not stimulate all or only the α1-receptors to which the endogenous amine has access as has been observed previously (Yang and Chiba, 2002). A second likely factor may be related to the fact, mentioned above, that in addition to their motor activating functions, brain α1- (and β1-)adrenoceptors in other regions play major roles in vigilance, fear and anxiety, behavioral processes that are frequently associated with inhibition of motor activity, particularly rearing behavior (van Dijken et al, 1992). Global brain activation of these receptors by high doses of ivt. EPI may therefore trigger both behavioral activation and inhibition, which was observed in the above experiments at high doses. The findings that α1-receptors in the hippocampus and septum suppress rather than stimulate active behavior together with the finding that coinfusion of the β1-receptor blocker, betaxolol, with ivt. EPI restored rearing in PNMT-inhibited animals strongly support this explanation. Therefore in order to evaluate these factors future research will need to study the behavioral actions of endogenous brain EPI release in the absence and presence of β-receptor antagonists at brain sites containing motoric α1-receptors vs sites containing other α1-receptors.
An additional problem for this hypothesis stems from the fact that deficiencies of dopamine-β-hydroxylase in humans and mice that totally deplete NE and EPI from the organism produce relatively subtle changes in behavior. Patients with this disorder have been reported to be apathetic and incapable of sustained exercise, but otherwise normal (Man in't Veld et al, 1987). DBH-deficient mice show an impairment of motor activity in swimming and rotarod tests, but no impairment of home-cage activity (Thomas and Palmiter, 1997). However, since these deficiencies exist throughout development, the high plasticity of the developing nervous system may compensate for impaired behavioral activity.
How the putative EPI-α1 system would be related to other adrenergic receptors and to the brain noradrenergic systems remains to be clarified. As discussed above, EPI is also an established neurotransmitter at central α2-adrenoceptors. The activation of CNS α2-receptors is well known to inhibit active behavior (Niittykoski et al, 1998), suggesting that brain EPI may have bidirectional control of activity via its effects on the two types of α-receptors. In this regard, it is of interest that a recent study has shown that α1- and α2-receptors are located on different cholinergic pontomesencephalic neurons suggesting that their afferent inputs may be separable (Hou et al, 2002). Also as noted above, the noradrenergic system may innervate those α1- and β1-adrenoceptors in forebrain regions related to arousal, vigilance, and anxiety based on the finding that selective destruction of noradrenergic neurons produces an upregulation of these two receptors in the hippocampus and neocortex (Reader and Briere, 1983). As noted above, since some degree of arousal is necessary for behavioral activation to occur, some degree of noradrenergic stimulation of these other α1-receptors would be necessary to permit behavioral activation by the putative EPI-α1-system. High noradrenergic activity in these areas, however, which occurs during severe anxiety, may suppress behavioral activation (Weiss et al, 1996; Weiss et al, 1998).
EFFECTS OF STRESS AND DEPRESSION ON EPI-α1-SYSTEM
At present, most studies point toward a marked impairment or inhibition of this system in depressives or in animal models of stress-induced depression.
With regard to presynaptic neurotransmitter function, a basic study in rats showed that hypothalamic EPI levels are significantly depleted by approximately 35% following an acute severe stress (3 min swim at 4°C) and gradually recover over the following 48 h. In this regard, it is of interest that the learned helplessness model of depression also requires 48 h to recover (Maier, 2001). Whether the latter depletion is severe enough to reduce EPI release in the brain has not yet been determined, although it is known that pharmacological inhibition of PNMT completely depletes extracellular EPI with only a 50% depletion of tissue levels (Routledge and Marsden, 1987b).
Only three small clinical studies have been performed on CSF EPI levels in depressed patients. The first (Berger et al, 1984) showed a large 70% reduction in depressed patients (0.017±0.008 nmol/ml) compared to ‘nuclear’ schizophrenics (those with hebephrenia or catatonia) (0.058±0.019 nmol/ml); however, a second control group consisting of other types of schizophrenics was found to have undetectable levels of the catecholamine. The second and third studies (Christensen et al, 1980; Gjerris et al, 1981) again showed approximately 75% reductions in EPI compared to neurological control groups. Moreover, there was a greater than four-fold rise in their CSF EPI levels after recovery from depression. If these results can be confirmed with larger samples, they would indicate a severe impairment or inhibition of central EPI release in depression.
At the level of the postsynaptic receptor most basic research has indicated that repeated stress and stress-related factors such as ACTH, corticosterone, and cytokine release can desensitize brain and peripheral α1-receptors, although some studies have suggested the reverse effect, that is, sensitization. We originally found that repeated footshock, restraint stress, ACTH, or corticosterone administration to rats reduced the α1-receptor-stimulated potentiation of cAMP responses in slices of the rat cerebral cortex and hypothalamus (Stone, 1979). These findings were confirmed by three other groups (Duman et al, 1985; Gannon and McEwen, 1990; Izumi et al, 1996), one of which showed that cotreatment with the antidepressant citalopram (but not imipramine) reversed this desensitization and also reversed the reduced behavioral activity that accompanied it (Izumi et al, 1997a, 1997b). However, repeated stress in rats was not found by a different group to reduce the α1-receptor-elicited phosphatidylinositol hydrolysis response to NE in slices of the cerebral cortex (Morinobu et al, 1992). In a more recent preliminary study, stress has been shown to abolish the α1-receptor-stimulated increase of GABA release in the rat amygdala in vivo (Braga et al, 2002). The foregoing changes in α1-responses may involve uncoupling of the receptor from its G-protein since most studies have failed to find a downregulation of α1-recognition sites in the brains of stressed animals (Lynch et al, 1983; Ossowska et al, 2001). However, it has been found that repeated restraint in rats markedly depletes the midbrain of α1-receptor mRNA (Miyahara et al, 1999), suggesting that some downregulation of the receptor may eventually occur.
Similar desensitizing effects of stress on α1-receptors were reported in the peripheral nervous system, where chronic stress reduced the α1-stimulated contraction of the isolated vas deferens (Singh et al, 2001) and downregulated the density of α1-binding sites in the heart (Torda et al, 1985). Furthermore, endotoxin (lipopolysaccharide, LPS) and septic shock markedly desensitized and downregulated vascular α1-receptors (Suba et al, 1992; Wakabayashi et al, 1989). Proinflammatory cytokine release is considered a likely etiological factor in depression (Anisman et al, 2002; Konsman et al, 2002).
While the above demonstrations of desensitization were intriguing, there were no basic studies on the effects of stress on the behavioral response to brain motoric α1-receptor stimulation. To investigate this our group examined the behavioral response to the stimulant, modafinil, in mice subjected to acute and repeated restraint stress (Stone et al, 2002b). Modafinil, as described above, stimulates prolonged motor activity that can be blocked by peripheral prazosin or ivt. terazosin. It was found that mice given three or more daily sessions of stress showed a reduced motor response to the drug. This change appeared to be selective for α1-receptors as the motor responses to dopaminergic agonists were not diminished and, in fact, were somewhat increased, in confirmation of previous findings by others (Hahn et al, 1986). In a preliminary experiment, we have also found that acute treatment with the cytokine releaser, LPS, transiently blocks the motor activity response to modafinil (Zhang et al, 2000).
Whether or not modafinil is an agonist at α1-receptors is still unclear. The compound does not activate and may inhibit phosphatidylinositol hydrolysis in mouse cortical brain slices yet it does stimulate a prazosin- or terazosin-blockable phosphorylation of MAP kinase (MAPK) both in vitro and in vivo but only at fairly high concentrations (10−4 M) (Stone et al, 2001d). Since it was the only (presumed) α1-agonist used in the above study, the possibility that stress may have acted on other neuronal systems necessary for modafinil action such as DA, orexin, or GABA (Ferraro et al, 1997; Scammell et al, 2000) cannot yet be excluded.
With regard to clinical findings, several studies are consistent with a reduced responsiveness of brain α1-receptors in depression. Checkley and Crammer (1977) and Asnis et al (1992) provided evidence for desensitized brain α1-receptors in depressed patients based on reductions in the plasma cortisol responses to amphetamine or DMI challenge, respectively. These responses had been shown to be mediated by α1-receptor stimulation (Laakman et al, 1986). With regard to [3H]prazosin-binding sites, Gross-Isseroff et al (1990) reported a significant downregulation in the tail of the corpus striatum in depressed suicides, whereas other brain regions showed no changes. De Paermentier et al (1997) also found no difference in the cortex of depressive suicides but Arango et al (1993) found increases in cortical α1-receptor density.
Basic studies on the effects of antidepressants and electroconvulsive shock on brain α1-receptors indirectly support the notion of an impaired function of α1-receptors in depression. Many authors have reported that chronic treatment with tricyclic antidepressants or ECS increases either the density (Maj et al, 1985; Nowak et al, 1988; Vetulani et al, 1984), agonist affinity (Maj et al, 2000; Menkes et al, 1983a), electrophysiological response (Menkes et al, 1980), or behavioral response of brain α1-receptors (Maj et al, 1998, 2000; Menkes et al, 1983b; Mogilnicka et al, 1987; Plaznik et al, 1984). As noted above, an antidepressant, citalopram, was also shown to reverse the stress-induced desensitization of the α1-potentiation of cAMP responses. However, most of these studies examined the cerebral cortex or hippocampus, which may predominantly contain α1-receptors related to vigilance and anxiety (Arnsten et al, 1999). Furthermore, not all investigators have been able to confirm the above findings (Heal, 1984; Stockmeier et al, 1987) indicating that the variables involved have not yet been completely isolated.
Not all basic and clinical research on depression, however, is in accord with α1-receptor desensitization. Harro and Oreland (2001) have recently suggested that catecholamine receptors in depressives may be sensitized by decreases in brain catecholamine release and therefore may show exaggerated responses to stress or drugs. In support of this notion, basic studies in mice and rats have shown that PNMT inhibitors produce a rapid sensitization (1–3 h) of animals to motor activation by i.p. amphetamine and dopa, and by ivt. α1-agonists (Katz et al, 1978, 1980; Stone et al, in press). The low CSF EPI levels found in depressives would be expected to sensitize α1-receptors. In addition, as noted above, corticosterone, which may be elevated during depressive illness, is essential for the expression of α1B-adrenoceptors in cell culture. Moreover, an increased euphoric effect of amphetamine has been found in depressed patients (Tremblay et al, 2002), while increases in the self-administration of amphetamine has been shown in an animal model of depression (Holmes et al, 2002). These changes could, of course, reflect sensitized dopaminergic receptors.
Resolving the above sensitization–desensitization controversy should be a primary goal of future clinical research in this area. A variety of factors may be at work here including the subtype and CNS location of α1-receptors, the durations of stress, hormone exposure and phase of illness, and possible alterations in the synergistic interactions with dopaminergic receptors. This work would be facilitated by a more definitive characterization of the action of modafinil at brain α1-receptors as this could potentially provide researchers with a clinical probe for central α1-receptor function. It would also be greatly aided by the development of clinical imaging techniques for studies of the density, occupancy, and coupling of brain α1-receptor subtypes in discrete brain regions concerned with the regulation of motor, positive motivation, and anxiety in these patients.
TROPHIC ACTIONS OF α1-RECEPTORS
Neurotransmission can produce long-term biochemical changes as well as short-term behavioral responses. With respect to the former, adrenergic receptors in the autonomic nervous system are believed to play a major role in long-term adaptation to stress by stimulating trophic, hyperplastic, and other long-term biochemical and morphological changes in end organs that enable adaptive changes in output (Ikeda et al, 1991; Stone, 1983). It has been proposed that these receptors trigger similar changes in the CNS during successful adaptation to chronic stress and antidepressant therapy to yield adaptive changes in neural output or plasticity brought about by actions of growth factors (Stone, 1983; Duman et al, 1999, 2000). In support of this, α1-receptor activation has been shown to stimulate both the expression of immediate early genes in the cortex, limbic system and brainstem during stress (Bing et al, 1991; Stone and Zhang, 1995) as well as the phosphorylation of cortical MAPK (Williams et al, 1998), which is involved in growth factor signaling processes. As noted above, in preliminary experiments, modafinil has been found to activate MAPK in cell culture as well as in vivo in the mouse cerebral cortex and to do so via stimulation of α1-receptors. It has also been found that administration of an α2-antagonist, yohimbine, which facilitates the release of brain catecholamines, increases the expression of nerve growth factor mRNA in the rat cortex (Stone et al, 1994), while catecholamines and/or indoleamines in cell culture (Ohta et al, 2002) as well as electroconvulsive shock and antidepressant drugs in vivo (Duman et al, 2000) can induce the expression of BDNF, GDNF, and/or NT3 as well as elicit functional and neuroanatomical signs of neural plasticity (D'Sa and Duman, 2002).
If the EPI-α1-system is impaired in depression, as we have proposed, then this may be a factor not only in the reduced behavioral activity of depressives but also in the neuronal atrophy and loss of glial cells in the prefrontal cortex and hippocampus, respectively, that occur in these patients (Drevets, 2000; McEwen, 2000). It should be noted that the human dentate gyrus has a very high concentration of α1-receptors (Palacios et al, 1987). However, it has been found recently that overexpression of constitutively active mutant α1-receptors causes neuronal degeneration in the mouse brain (Zuscik et al, 2000) indicating the need for careful dose–response studies to avoid excessive stimulation. Further research will be necessary to define the mechanism of these changes and to determine how they are related to long-term changes in behavior.
SUMMARY
New behavioral and neuropharmacological evidence has implicated a subgroup of brain α1B-adrenoceptors as a key factor in positively motivated behavioral activity. Most of these ‘motoric’ α1-receptors are located in or close to monoamine-containing neuron cell bodies (NE and 5HT) or their terminal targets (nucleus accumbens—DA) and appear to receive EPI as their neurotransmitter. It is speculated that this ‘EPI-innervated-α1-system’ activates behavior by producing a coordinated excitation of the major monoaminergic systems of the brain. There is evidence that this system is impaired or inhibited in depressive illness from the findings of low levels of EPI in the CSF and of altered responsiveness of brain α1-receptors in depressed patients. There is also evidence that its impairment may facilitate CNS brain atrophic effects in depression as it is linked to growth factor induction and MAPK activation. As a number of antidepressant agents are capable of restoring or enhancing its function, the EPI-α1-system would appear to represent a new target for this illness.
Further research is needed on many facets of the above putative system including its relation to aversive as opposed to appetitive behavioral activation, its relation to C1 and C2 medullary EPI-containing neurons as opposed to brain cells containing only PNMT, its status in depression in larger clinical studies, and further basic science studies of the effects of reuptake inhibitors, stress, and immune factors on its function.
References
Acosta-Martinez M, Fiber JM, Brown RD, Etgen AM (1999). Localization of α1B-adrenergic receptor in female rat brain regions involved in stress and neuroendocrine function. Neurochem Int 35: 383–391.
Anden NE, Strombom U, Svensson TH (1973). Dopamine and noradrenaline receptor stimulation: reversal of reserpine-induced suppression of motor activity. Psychopharmacologia 29: 289–298.
Anisman H, Hayley S, Turrin N, Merali Z (2002). Cytokines as a stressor: implications for depressive illness. Int J Neuropsychopharmacol 5: 357–373.
Arango V, Ernsberger P, Sved A, Mann J (1993). Qauntitative autoradiography of α1- and α2-adrenergic receptors in the cerebral cortex of controls and suicide victims. Brain Res 630: 271–282.
Arnsten AFT, Mathew R, Ubriani R, Taylor JR, Bao-Ming L (1999). α-1 Noradrenergic receptor stimulation impairs prefrontal cortical cognitive function. Biol Psychiatry 45: 26–31.
Asnis GM, Sanderson WC, van Praag HM (1992). Cortisol response to intramuscular desipramine in patients with major depression and normal control subjects: a replication study. Psychiatry Res 44: 237–250.
Aston-Jones G, Astier B, Ennis M (1992). Inhibition of noradrenergic locus coeruleus neurons by C1 adrenergic cells in the rostral ventral medulla. Neuroscience 48: 371–381.
Aston-Jones G, Rajkowski J, Kubiak P, Alexinsky T (1994). Locus coeruleus neurons in monkey are selectively activated by attended cues in a vigilance task. J Neurosci 14: 4467–4480.
Astrachan D, Davis M, Gallager D (1983a). Behavior and binding: correlations between α1-adrenergic stimulation of acoustic startle and α1-adrenoceptor occupancy and number in rat lumbar spinal cord. Brain Res 260: 81–90.
Astrachan D, Gallager D, Davis M (1983b). Behavior and binding: desensitization to α1-adrenergic stimulation of acoustic startle is associated with a decrease in α1-adrenoceptor binding sites. Brain Res 276: 183–187.
Auclair A, Cotecchia S, Glowinski J, Tassin JP (2002). D-amphetamine fails to increase extracellular dopamine levels in mice lacking alpha 1b-adrenergic receptors: relationship between functional and nonfunctional dopamine release. J Neurosci 22: 9150–9154.
Bakshi VP, Geyer MA (1999). Alpha-1-adrenergic receptors mediate sensorimotor gating deficits produced by intracerebral dizocilpine administration in rats. Neuroscience 92: 113–121.
Baraban JM, Aghajanian G (1980). Suppression of firing activity of 5-HT neurons in the dorsal raphe by alpha-adrenoceptor antagonists. Neuropharmacology 19: 355–363.
Berger P, King RA, Lemoine C, Mefford I (1984). Cerebrospinal fluid epinephrine concentrations: discrimination of subtypes of depression and schizophrenia. Psychopharmacol Bull 20: 412–415.
Berretta N, Bernardi G, Mercuri NB (2000). Alpha1-adrenoceptor-mediated excitation of substantia nigra pars reticulata neurons. Neuroscience 98: 599–604.
Bing G, Filer D, Miller JC, Stone EA (1991). Noradrenergic activation of immediate early genes in rat cerebral cortex. Mol Brain Res 11: 43–46.
Blanc G, Trovero F, Vezina P, Herve D, Godeheu A-M, Glowinski J et al (1994). Blockade of prefronto-cortical α1-adrenergic receptors prevents locomotor hyperactivity induced by subcortical D-amphetamine injection. Eur J Neurosci 6: 293–298.
Braga MFM, Hough CJ, Manion S, Li H (2002). Chronic stress causes impairment of 1-adrenoceptor-mediated modulation of GABAergic synaptic transmission in the basolateral amygdala. Abstr Soc Neurosci 28, Prog #103.10.
Brown AS, Gershon S (1993). Dopamine and depression. J Neural Transm 91: 75–109.
Cabib S, Giardino L, Calza L, Zanni M, Mele A, Puglisi-Allegra S (1998). Stress promotes major changes in dopamine receptor densities within the mesoaccumbens and nigrostriatal systems. Neuroscience 84: 193–200.
Cabib S, Puglisi-Allegra S (1996). Stress, depression and the mesolimbic dopamine system. Psychopharmacology 128: 331–342.
Cano J, Garcia-Uria J, Machado A, Reinoso-Suarez F (1980). Effect of cerebellar lesions on monoamine levels in various brain areas of the cat. J Neurochem 35: 1446–1448.
Cavalli A, Lattion AL, Hummler E, Nenniger M, Pedrazzini T, Aubert JF et al (1997). Decreased blood pressure response in mice deficient of the α1b-adrenergic receptor. Proc Natl Acad Sci USA 94: 11589–11594.
Chamba G, Weissmann D, Rousset C, Renaud B, Pujol JF (1991). Distribution of alpha-1 and alpha-2 binding sites in the rat locus coeruleus. Brain Res Bull 26: 185–193.
Chan-Palay V (1977). Cerebellar Dentate Nucleus: Organization, Cytology and Transmitters. Springer: Berlin. pp 297–363.
Checkley S, Crammer J (1977). Hormone responses to methylamphetamine in depression. Br J Psychiatry 131: 582–586.
Chopde C, Hote M, Mandhane S, Muthal A (1995). Glucocorticoids attenuate haloperidol-induced catalepsy through adrenal catecholamines. J Neural Transm 102: 47–54.
Christensen NJ, Vestergaard P, Sorensen T, Rafaelsen OJ (1980). Cerebrospinal fluid adrenaline and noradrenaline in depressed patients. Acta Psychiatr Scand 61: 178–182.
Chu HP, Etgen AM (1999). Ovarian hormone dependence of α1-adrenoceptor activation of the nitric oxide-cGMP pathway: relevance for hormonal facilitation of lordosis behavior. J Neurosci 19: 7191–7197.
Cools A (1991). Differential role of mineralocorticoid and glucocorticoid receptors in the genesis of desamphetamine-induced sensitization of mesolibmic, α1 adrenergic receptors in the ventral striatum. Neuroscience 43: 419–428.
D'Sa C, Duman R (2002). Antidepressants and neuroplasticity. Bipolar Disord 4: 183–194.
Day HEW, Campeau S, Watson Jr SJ, Akil H (1997). Distribution of α1a-, α1b- and α1d-adrenergic receptor mRNA in the rat brain and spinal cord. J Chem Neuroanat 13: 115–139.
De Paermentier F, Mauger JM, Lowther S, Crompton MR, Katona CL, Horton RW (1997). Brain alpha-adrenoceptors in depressed suicides. Brain Res 757: 60–68.
Donaldson IM, Dolphin A, Jenner P, Marsden CD, Pycock C (1976). Contraversive circling behaviour produced by unilateral electrolytic lesions of the ventral noradrenergic bundle mimicking the changes seen with unilateral electrolytic lesions of the locus coeruleus. J Pharm Pharmacol 28: 329–331.
Drevets WC (2000). Neuroimaging studies of mood disorders. Biol Psychiatry 48: 813–829.
Drouin C, Darracq L, Trovero F, Blanc G, Glowinski J, Cotecchia S et al (2002). α1b-Adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J Neurosci 22: 2873–2884.
Dubocovich ML, Mogilnicka E, Areso PM (1990). Antidepressant-like activity of the melatonin receptor antagonist, luzindole(N-0774), in the mouse behavioral despair test. Eur J Pharmacol 182: 313–325.
Duman RS, Malberg J, Nakagawa S, D'Sa C (2000). Neuronal plasticity and survival in mood disorders. Biol Psychiatry 48: 732–739.
Duman RS, Malberg J, Thome J (1999). Neural plasticity to stress and antidepressant treatment. Biol Psychiatry 46: 1181–1191.
Duman RS, Strada SJ, Enna SJ (1985). Effect of imipramine and adrenocorticotropin administration on the rat brain norepinephrine-coupled cyclic nucleotide generating system: alterations in alpha and beta adrenergic component. J Pharmacol Exp Ther 234: 409–414.
Duteil J, Rambert FA, Pessonnier J, Hermant J-F, Gombert R, Assous E (1990). Central α1-adrenergic stimulation in relation to the behaviour stimulating effect of modafinil; studies with experimental animals. Eur J Pharmacol 180: 49–58.
Engberg G, Elam M, Svensson T (1981). Effect of adrenaline synthesis inhibition on brain noradrenaline neurons in locus coeruleus. Brain Res 223: 49–58.
Eshel G, Ross SB, Kelder D, Edis LE, Jackson DM (1990). 1 (but not α2)-adrenoreceptor agonists in combination with the dopamine D2 agonist quinpirole produce locomotor stimulation in dopamine-depleted mice. Pharmacol Toxicol 67: 123–131.
Espejo EF, Miñano J (2001). Adrenergic hyperactivity and metanephrine excess in the nucleus accumbens after prefrontocortical dopamine depletion. J Neurophysiol 85: 1270–1274.
Ferraro L, Antonelli T, O'Connor WT, Tanganelli S, Rambert FA, Fuxe K (1997). Modafinil: an antinarcoleptic drug with a different neurochemical profile to D-amphetamine and dopamine uptake blockers. Biol Psychiatry 42: 1181–1183.
Ferry B, Roozendaal B, McGaugh JL (1999). Role of norepinephrine in mediating stress hormone regulation of long-term memory storage: a critical involvement of the amygdala. Biol Psychiatry 46: 1140–1152.
Feuvrier E, Aubert M, Mauss AL, Alonso G, Gaillet S, Malaval F et al (1998). Glucocorticoids provoke a shift from α2- to α1-adrenoreceptor activities in cultured hypothalamic slices leading to opposite noradrenaline effect on corticotropin-releasing hormone release. J Neurochem 70: 1199–1209.
Fujimaki K, Morinobu S, Duman R (2000). Administration of a cAMP phosphodiesterase 4 inhibitor enhances antidepressant-induction of BDNF mRNA in rat hippocampus. Neuropsychopharmacology 22: 42–51.
Fuller R (1982). Pharmacology of brain epinephrine neurons. Ann Rev Pharmacol Toxicol 22: 31–55.
Gannon MN, McEwen BS (1990). Calmodulin involvement in stress- and corticosterone-induced down-regulation of cyclic AMP-generating systems in brain. J Neurochem 55: 276–284.
Garcia-Sainz J, Vazquez-Prado J, Villalobos-Molina R (1999). α1-Adrenoceptors: subtypes, signaling, and roles in health and disease. Arch Med Res 30: 449–458.
Geyer MA, Segal DS, Mandell AJ (1972). Effect of intraventricular infusion of dopamine and norepinephrine on motor activity. Physiol Behav 8: 653–658.
Gioanni Y, Thierry AM, Glowinski J, Tassin JP (1998). α1-Adrenergic, D1, and D2 receptors interactions in the prefrontal cortex: implications for the modality of action of different types of neuroleptics. Synapse 30: 362–370.
Gjerris A, Jensen EW, Christensen NJ, Rafaelsen OJ (1981). Adrenaline and noradrenaline in psychiatric disorders. In: Perris C, Struwe G, Jansson B (eds) Biological Psychiatry. Elsevier: Copenhagen. pp 565–568.
Grenhoff J, Nisell M, Ferre S, Aston-Jones G, Svensson TH (1993). Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J Neural Transm 93: 11–25.
Grenhoff J, North RA, Johnson SW (1995). α1-Adrenergic effects on dopamine neurons recorded intracellularly in the rat midbrain slice. Eur J Neurosci 7: 1707–1713.
Gross-Isseroff R, Dillon KA, Fieldust SJ, Biegon A (1990). Autoradiographic analysis of α1-noradrenergic receptors in the human brain postmortem. Arch Gen Psychiatry 47: 1049–1053.
Hahn B, Zacharko R, Anisman H (1986). Alterations of amphetamine elicited perseveration and locomotor excitation following acute and repeated stressor application. Pharmacol Biochem Behav 25: 29–33.
Harro J, Oreland L (2001). Depression as a spreading adjustment disorder of monoaminergic neurons: a case for primary implication of the locus coeruleus. Brain Res Rev 38: 79–128.
Hasegawa K (1975). Changes in the self-stimulation behavior by intraventricular injection of epinephrine, norepinephrine, isoproterenol and dopamine. Jpn J Pharmacol 25: 616–619.
Heal DJ (1984). Phenylephrine-induced activity in mice as a model of central α1-adrenoceptor function. Effects of acute and repeated administration of antidepressant drugs and electroconvulsive shock. Neuropharmacology 23: 1241–1251.
Herman JP, Stinus L, LeMoal M (1984). Repeated stress increases locomotor response to amphetamine. Psychopharmacology 84: 431–435.
Hieble JP, Kolpak DC, McCafferty GP, Ruffolo Jr RR, Testa R, Leonardi A (1999). Effects of α1-adrenoceptor antagonists on agonist and tilt-induced changes in blood pressure: relationships to uroselectivity. Eur J Pharmacol 373: 51–62.
Hokfelt T, Johansson O, Goldstein M (1984). Central catecholamine neurons as revealed by immunohistochemistry with special reference to adrenaline neurons. In: Bjorklund A, Hokfelt T (eds) Handbook of Chemical Neuroanatomy. Elsevier: New York. pp 157–276.
Holmes P, Masini C, Primeaux S, Garrett J, Zellner A, Stogner K et al (2002). Intravenous self-administration of amphetamine is increased in a rat model of depression. Synapse 46: 4–10.
Hou Y, Manns I, Jones B (2002). Immunostaining of cholinergic pontomesencephalic neurons for α1 versus α2 adrenergic receptor suggests different sleep–wake state activities and roles. Neuroscience 114: 517–521.
Ikeda U, Tsuruya Y, Yaginuma T (1991). α1-Adrenergic stimulation is coupled to cardiac myocyte hypertrophy. Am J Physiol 260: H953–H956.
Izumi J, Washizuka M, Hayashi-Kuwabara Y, Yoshinaga K, Tanaka Y, Ikeda Y et al (1996). An attenuated α1-potentiation of β-adrenoceptor-stimulated cyclic AMP formation after repeated saline injections in Fischer 344 strain rats. Life Sci 59: 33–42.
Izumi J, Washizuka M, Hayashi-Kuwabara Y, Yoshinaga K, Tanaka Y, Ikeda Y et al (1997a). Protective effect of citalopram against the attenuation of the alpha-1-potentiation of cAMP formation in Fischer 344 strain rats. Behav Brain Res 83: 209–212.
Izumi J, Washizuka M, Kuwabara YH, Yoshinaga K, Tanaka Y, Ikeda Y et al (1997b). Evidence for a depressive-like state induced by repeated saline injections in Fisher 344 rats. Pharmacol Biochem Behav 57: 883–888.
Johnson RD, Minneman KP (1986). Characterization of α1-adrenoceptors which increase cyclic AMP accumulation in rat cerebral cortex. Eur J Pharmacol 129: 293–305.
Jones LS, Gauger LL, Davis JN (1985). Anatomy of brain alpha-1 adrenergic receptors: in vitro autoradiography with [125I]-heat. J Comp Neurol 231: 190–208.
Katz R, Carroll B (1978). Inhibition of phenylethanolamine-N-methyltransferase and brain-stimulated reward. Psychopharmacology 57: 39–42.
Katz R, Carroll B, Leibler L (1978). Enhancement of drug-induced motor activity by an inhibitor of phenylethanol-N-methyltransferase. Neurosci Lett 8: 83–88.
Katz R, Gelbart J, Carroll B (1980). Potentiation of l-dopa induced motor activity by an inhibitor of phenylethanolamine-N-methyltransferase. Prog Neuropsychopharmacol 4: 101–105.
Kelsey J (1976). Behavioral effects of intraseptal injections of adrenergic drugs in rats. Physiol Psychol 4: 433–438.
Kleinrok Z, Zebrowska-Lupina I (1971). Central action of phentolamine administered intraventricularly in the rat. Psychopharmacology 20: 348–354.
Knauber J, Muller W (2000). Decreased exploratory activity and impaired passive avoidance behaviour in mice deficient for the α1b-adrenoceptor. Eur Neuropsychopharmacol 10: 423–427.
Konsman JP, Parnet P, Dantzer R (2002). Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci 25: 154–159.
Kostowski W, Jerlicz M, Bidzinski A, Hauptmann M (1978). Evidence for the existence of two opposite noradrenergic brain systems controlling behavior. Psychopharmacology 59: 311–312.
Laakman G, Wittmann M, Schoen M, Zugan K, Weiss A, Meissner R et al (1986). Effects of receptor blockers (methysergide, propranolol, phentolamine, yohimbine and prazosin) on desipramine-induced pituitary hormone stimulation in humans. Psychoneuroendocrinology 11: 475–489.
Lin Y, Stone EA (2001). Effect of local blockade of alpha 1-adrenoceptors in various brain regions on locomotor activity and movement in mice. Abstr Soc Neurosci 27, Prog #292.4.
Lin Y, Stone EA, Ahsan R, Quartermain D (2002). Antagonist mapping of the mouse brain for alpha-1-adrenoceptors that regulate motor activity. Abstr Soc Neurosci 28, Prog #879.8.
Lindroos O, Leinonen L, Hilakivi I (1984). Autoradiographic picture of mouse brain after intraperitoneal injection of [3H]prazosin. Acta Universit Tamper 21: 131–141.
Lipinski JF, Cohen BM, Zubenko GS, Waternaux C (1987). Adrenoceptors and the pharmacology of affective illness: a unifying theory. Life Sci 40: 1947–1963.
Lynch M, Littelton J, McKernan R, Durcan M, McMillan T, Campbell I (1983). Adrenoceptor number and function in rat cortex after ethanol and immobilization stress. Brain Res 288: 145–149.
MacDonald E, Sirvio J (1999). Central α1-adrenoceptors: their role in the modulation of attention and memory formation. Pharmacol Ther 83: 49–65.
Maier SF (2001). Exposure to the stressor environment prevents the temporal dissipation of behavioral depression/learned helplessness. Biol Psychiatry 49: 763–773.
Maj J, Klimek V, Nowak G (1985). Antidepressant drugs given repeatedly increase binding to alpha 1-adrenoceptors in the rat cortex. Eur J Pharmacol 119: 113–116.
Maj J, Rogóz Z, Dlaboga D, Dziedzicka-Wasylewska M (2000). Pharmacological effects of milnacipran, a new antidepressant, given repeatedly on the α1-adrenergic and serotonergic 5-HT2A systems. J Neural Transm 107: 1345–1359.
Maj J, Rogóz Z, Skuza G, Margas W (1998). Repeated trimipramine induces dopamine D2/D3 and α1-adrenergic up-regulation. J Neural Transm 105: 329–342.
Man in't Veld AJ, Moleman P, Boomsma F, Schalekamp MADH (1987). Congenital dopamine-beta-hydroxylase deficiency. A novel orthostatic syndrome. Lancet 1: 183–187.
Mason ST, Roberts DC, Fibiger HC (1978). Noradrenergic influences on catalepsy. Psychopharmacology 60: 53–57.
Mathé JM, Nomikos GG, Hildebrand BE, Hertel P, Svensson TH (1996). Prazosin inhibits MK-801-induced hyperlocomotion and dopamine release in the nucleus accumbens. Eur J Pharmacol 309: 1–11.
McEwen BS (2000). Effects of adverse experiences for brain structure and function. Biol Psychiatry 48: 721–731.
McNaughton N, Mason ST (1980). The neuropsychology and neuropharmacology of the dorsal ascending noradrenergic bundle—a review. Prog Neurobiol 14: 157–219.
Mefford I (1988). Epinephrine in mammalian brain. Prog Neuropsychopharmacol 12: 365–388.
Mefford IN (1987). Are there epinephrine neurons in rat brain? Brain Res Rev 12: 383–395.
Menkes DB, Aghajanian G, McCall R (1980). Chronic antidepressant treatment enhances α-adrenergic and serotonergic responses in the facial nucleus. Life Sci 27: 45–55.
Menkes DB, Aghajanian GK, Gallager DW (1983a). Chronic antidepressant treatment enhances agonist affinity of brain α1-adrenoceptors. Eur J Pharmacol 87: 35–41.
Menkes DB, Baraban JM, Aghajanian G (1981). Prazosin selectively antagonizes neuronal responses mediated by alpha-1 adrenoceptors in brain. Naunyn-Schmiedebergs Arch Pharmacol 317: 273–275.
Menkes DB, Kehne J, Gallager W, Aghajanian G, Davis M (1983b). Functional supersensitivity of CNS α1-adrenoceptors following chronic antidepressant treatment. Life Sci 33: 181–188.
Miyahara S, Komori T, Fujiwara R, Shizuya K, Yamamoto M, Ohmori M et al (1999). Effects of single and repeated stresses on the expression of mRNA for or α1-adrenoceptors in the rat hypothalamus and midbrain. Eur J Pharmacol 379: 111–114.
Mogilnicka E, Zazula M, Wedzony K (1987). Functional supersensitivity to the α1-adrenoceptor agonist after repeated treatment with antidepressant drugs is not conditioned by B-down-regulation. Neuropharmacology 26: 1457–1461.
Morin D, Sapena R, Zini R, Onteniente B, Tillement JP (1996). Characterization of β-adrenergic receptors of freshly isolated astrocytes and neurons from rat brain. Life Sci 60: 315–324.
Morinobu S, Kuwayama N, Kawanami T, Okuyama N, Takahashi M, Totsuka S et al (1992). Influence of the acute stress on agonist-stimulated phosphinositide hydrolysis in the rat cerebral cortex. Prog Neuropsychopharmacol Biol Psychiatry 16: 561–570.
Murrough J, Boss-Williams K, Emery M, Bonsall R, Weiss J (2000). Depletion of brain norepinephrine does not reduce spontaneous ambulatory activity of rats in the home cage. Brain Res 883: 125–130.
Nakamura S, Sakaguchi T, Kimura F, Aoki F (1988). The role of alpha1-adrenoceptor-mediated collateral excitation in the regulation of the electrical activity of locus coeruleus neurons. Neuroscience 27: 921–929.
Nemeroff C (2002). Recent advances in the neurobiology of depression. Psychopharmacol Bull 36: 6–23.
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002). Neurobiology of depression. Neuron 34: 13–25.
Niittykoski M, Lappalainen R, Jolkkonen J, Haapalinna A, Riekkinen Sr P, Sirvio J (1998). Systemic administration of atipamezole, a selective antagonist of alpha-2 adrenoceptors, facilitates behavioural activity but does not influence short-term or long-term memory in trimethyltin-intoxicated and control rats. Neurosci Biobehav Rev 22: 735–750.
Nishino S, Fruhstorfer B, Arrigoni J, Guilleminault C, Dement WC, Mignot E (1993). Further characterization of the alpha-1 receptor subtype involved in the control of cataplexy in canine narcolepsy. J Pharmacol Exp Ther 264: 1079–1084.
Nowak JZ, Przybysz M, Zurawska E (1988). The melatonin generating system in the rat retina and pineal gland: effect of single and repeated electroconvulsive shock (ECS). Pol J Pharmacol Pharm 40: 573–584.
Ohliger-Frerking P, Horowitz J, Horwitz B (2002). Enhanced adrenergic excitation of serotonergic dorsal raphe neurons in genetically obese rats. Neurosci Lett 332: 107–110.
Ohta K, Ohta M, Mizuta I, Fujinami A, Shimazu S, Sato N et al (2002). The novel catecholaminergic and serotoninergic activity enhancer R-(−)-1-(benzofuran-2-yl)-2-propylaminopentane up-regulates neurotrophic factor synthesis in mouse astrocytes. Neurosci Lett 328: 205–208.
Ossowska G, Nowak G, Kata R, Klenk-Majewska B, Danilczuk Z, Zebrowska-Lupina F (2001). Brain monoamine receptors in a chronic unpredictable stress model in rats. J Neural Transm 108: 311–319.
Palacios JM, Hoyer D, Cortes R (1987). α1-Adrenoceptors in the mammalian brain: similar pharmacology but different distributions in rodents and primates. Brain Res 419: 65–75.
Phillipu A, Dietl H, Sinha JN (1979). In vivo release of endogenous catecholamines in the hypothalamus. Naunyn Schmiedebergs Arch Pharmacol 308: 137–143.
Piao H, Taniguchi T, Nakamura S, Zhu J, Suzuki F, Mikami D et al (2000). Cloning of rabbit α1b-adrenoceptor and pharmacological comparison of α1a-, α1b-, and α1d-adrenoceptors in rabbit. Eur J Pharmacol 396: 9–17.
Plaznik A, Danysz W, Kostowski W (1984). Behavioral evidence for alpha-1 adrenoceptor up- and alpha-2-adrenoceptor down regulation in the rat hippocampus after chronic desipramine treatment. Eur J Pharmacol 101: 305–306.
Reader T, Briere R (1983). Long-term unilateral noradrenergic denervation: monoamine content and [3H]-prazosin binding sites in rat neocortex. Brain Res Bull 11: 687–692.
Reinhard Jr JF, Galloway MP, Roth R (1983). Noradrenergic modulation of serotonin synthesis and metabolism. II. Stimulation by 3-isobutyl-1-methylxanthine. J Pharmacol Exp Ther 226: 764–769.
Roth K, Mefford I, Barchas J (1982). Epinephrine, norepinephrine, dopamine and serotonin: differential effects of acute and chronic stress on regional brain amines. Brain Res 239: 417–424.
Rouqier I, Claustre Y, Benavidas J (1994). Alpha-1 adrenoceptor antagonists differentially control serotonin release in the hippocampus and striatum: a microdialysis study. Eur J Pharmacol 261: 59–64.
Routledge C, Marsden C (1987a). Adrenaline in the CNS: in vivo evidence for a functional pathway innervating the hypothalamus. Neuropharmacology 26: 823–830.
Routledge C, Marsden CA (1987b). Comparison of the effects of selected drugs on the release of hypothalamic adrenaline and noradrenaline measured in vivo comparison of the effects of selected drugs on the release of hypothalamic adrenaline and noradrenaline measured in vivo. Brain Res 426: 103–111.
Sakaue M, Hoffman BB (1991). Glucocorticoids induce transcription and expression of the alpha-1B adrenergic receptor gene in DTT1 MF-2 smooth muscle cells. J Clin Invest 88: 385–389.
Saldalge A, Coughlin L, Fu H, Wang B, Blendy J (2002). The role of αID adrenoceptors in modulating locomotor responses. Abstr Soc Neurosci 28, Prog #879.6.
Sauter A, Ueta K, Goldstein M (1980). Effect of DBH- and PNMT-inhibition on brain adrenaline and noradrenaline in animals exposed to stress. In: Fuxe K, Goldstein M, Hokfelt B, Hokfelt T (eds) Central Adrenaline Neurons. Pergamon Press: Oxford. pp 97–103.
Scammell TE, Estabrooke IV, McCarthy MT, Chemelli RM, Yanagisawa M, Miller MS et al (2000). Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci 20: 8620–8628.
Schwinn DA (2000). Novel role for α1-adrenergic receptor subtypes in lower urinary tract symptoms. Br J Urol Int 86: 11–22.
Segal D, Geyer M, Weiner B (1975). Strain differences during intraventricular infusion of norepinephrine: possible role of receptor sensitivity. Science 189: 301–303.
Segal DS, McAllister C, Geyer MA (1974). Ventricular infusion of norepinephrine and amphetamine: direct versus indirect action. Pharmacol Biochem Behav 2: 79–86.
Simon P, Hemet C, Ramassamy C, Costentin J (1995). Non-amphetaminic mechanism of stimulant locomotor effect of modafinil in mice. Eur Neuropsychopharmacol 5: 509–514.
Singh A, Einstein R, Lavidis N (2001). Effects of restraint stress on responsiveness of atria and vas deferens in Sprague–Dawley rats. J Autonom Pharmacol 21: 255–261.
Snider RS, Maiti A, Snider SR (1976). Cerebellar pathways to ventral midbrain and nigra. Exp Neurol 53: 714–728.
Snider SR, Snider RS (1979). Kainic acid: enduring alterations in cerebellar morphology and in cerebral catecholamine and GABA concentrations after cerebellar injection in the rat. Neurosci Lett 12: 339–342.
Spreng M, Cotecchia S, Schenk F (2001). A behavioral study of alpha-1b adrenergic receptor knockout mice: increased reaction to novelty and selectively reduced learning capacities. Neurobiol Learn Mem 75: 214–229.
Stevens D, McCarley R, Greene R (1994). The mechanism of noradrenergic α1 excitatory modulation of pontine reticular formation neurons. J Neurosci 14: 6481–6487.
Stockmeier CA, McLeskey SW, Blendy JA, Armstrong NR, Kellar KJ (1987). Electroconvulsive shock but not antidepressant drugs increases α1-adrenoceptor binding sites in rat brain. Eur J Pharmacol 139: 259–266.
Stone EA (1979). Reduction by stress of norepinephrine-stimulated accumulation of cyclic AMP in rat cerebral cortex. J Neurochem 32: 1335–1337.
Stone EA (1983). Problems with current catecholamine hypotheses of antidepressant drugs. Speculations leading to a new hypothesis. Behav Brain Sci 6: 535–578.
Stone EA, Cotecchia S, Lin Y, Quartermain D (2002a). Role of brain α1B-adrenoceptors in modafinil-induced behavioral activity. Synapse 46: 269–270.
Stone E, Grunewald G, Lin Y, Ahsan R, Rosengarten H, Kramer K et al (in press). Role of epinephrine stimulation of CNS α1-adrenoceptors in motor activity in mice. Synapse.
Stone EA, Kramer HK (2001a). Biphasic action of corticosterone on alpha 1B-adrenoceptors in DDT1 MF2 cells. FASEB J 15: A549.
Stone EA, Lin Y, Itteera A, Quartermain D (2001b). Pharmacological evidence for the role of brain alpha 1B-adrenoceptor activity in the motor activity and spontaneous movement in mice. Neuropharmacology 40: 254–261.
Stone E, Lin Y, Suckow R, Quartermain D (2002b). Stress-induced subsensitivity to modafinil and its prevention by corticosteroids. Pharmacol Biochem Behav 73: 971–978.
Stone EA, Zhang Y (1995). Adrenoceptor antagonists block c-fos response to stress in the mouse brain. Brain Res 694: 279–286.
Stone EA, Manavalan JS, Basham DA, Bing G (1994). Effect of yohimbine on nerve growth factor mRNA and protein levels in rat hippocampus. Neurosci Lett 167: 11–13.
Stone EA, Quartermain D (1999). Alpha 1-adrenergic neurotransmission, corticosterone and behavioral depression. Biol Psychiatry 46: 1287–1300.
Stone EA, Rosegarten H, Kramer HK (2001d). Modafinil has partial agonist activity at alpha 1B-adrenoceptors in vitro and in vivo. Abstr Soc Neurosci, 27, Prog #292.4.
Stone E, Rosengarten H, Lin Y, Quartermain D (2001c). Pharmacological blockade of brain alpha 1-adrenoceptors as measured by ex vivo [3H]prazosin binding is correlated with behavioral immobility. Eur J Pharmacol 420: 97–102.
Stone E, Zhang Y, Rosengarten H, Yeretsian J, Quartermain D (1999). Brain α1-adrenergic neurotransmission is necessary for behavioral activation to environmental change in mice. Neuroscience 94: 1245–1252.
Suba EA, McKenna TM, Williams TJ (1992). In vivo and in vitro effects of endotoxin on vascular responsiveness to norepinephrine and signal transduction in the rat. Circ Shock 36: 127–133.
Taylor JA, Twomey TM, Schach von Wittenau M (1977). The metabolic fate of prazosin. Xenobiotica 7: 357–364.
Thomas SA, Palmiter RD (1997). Disruption of the dopamine beta-hydroxylase gene in mice suggests roles for norepinephrine in motor function, learning and memory. Behav Neurosci 111: 579–589.
Torda T, Kvetnansky R, Petrikova M (1985). Effect of repeated immobilization stress on central and peripheral adrenoceptors in rats. Endo Exp 19: 157–163.
Tremblay LK, Naranjo CA, Cardenas L, Herrmann N, Busto UE (2002). Probing brain reward system function in major depressive disorder—altered response to dextroamphetamine. Arch Gen Psychiatry 59: 409–416.
Trovero F, Blanc G, Herve D, Vezina P, Glowinski J, Tassin J-P (1992). Contribution of an alpha1-adrenergic receptor subtype to the expression of the ‘ventral tegmental area syndrome’. Neuroscience 47: 69–76.
U'Prichard D, Loftus D, Stolk J (1988). Functional association between epinephrine-containing neurons and α-adrenoceptors in discrete brain areas: analysis in inbred rat strains. In: Stolk J, U'Prichard D, Fuxe K (eds) Epinephrine in the Central Nervous System. Oxford: New York. pp 195–211.
van Dijken HH, Mos J, Van Der Heyden JAM, Tilders FJH (1992). Characterization of stress-induced long-term behavioural changes in rats: evidence in favor of anxiety. Physiol Behav 52: 945–951.
Vantini G, Perry B, Guchhait R, U'Prichard D, Stolk J (1984). Brain epinephrine systems: detailed comparison of adrenergic and noradrenergic metabolism, receptor number and in vitro regulation, in two inbred rat strains. Brain Res 296: 49–65.
Vetulani J, Antkiewicz-Michaluk L, Rokosz-Pelc A (1984). Chronic administration of antidepressant drugs increases the density of cortical [3H]prazosin binding sites in the rat. Brain Res 310: 360–362.
Volgin D, Mackiewicz M, Kubin L (2001). 1B receptors are the main postsynaptic mediators of adrenergic excitation in brainstem motoneurons, a single-cell RT-PCR study. J Chem Neuroanat 22: 157–166.
Wakabayashi I, Hatake K, Kakishita E, Hishida S, Nagai K (1989). Desensitization of alpha-1 adrenergic receptor mediated smooth muscle contraction in aorta from endotoxic rats. Life Sci 45: 509–515.
Watson S, Gallagher P, Del-Estal D, Hearn A, Ferrier IN, Young AH (2002). Hypothalamic–pituitary–adrenal axis function in patients with chronic depression. Psychol Med 32: 1021–1028.
Weiss J, Bonsall RW, Demetrikopoulos MK, Emery MS, West CHK (1998). Galanin: a significant role in depression? Ann NY Acad Sci 863: 364–382.
Weiss J, Demetrikopoulos M, West C, Bonsall R (1996). Hypothesis linking the noradrenergic and dopaminergic systems in depression. Depression 3: 225–245.
Williams A, Morilak D (1997). α1B adrenoceptors in rat paraventricular nucleus overlap with, but do not mediate, the induction of c-fos expression by osmotic or restraint stress. Neuroscience 76: 901–913.
Williams J, Marshall K (1987). Membrane properties and adrenergic responses in locus coeruleus neurons of young rats. J Neurosci 7: 3687–3694.
Williams NG, Zhong H, Minneman KP (1998). Differential coupling of α1-, α2-, and β-adrenergic receptors to mitogen-activated protein kinase pathways and differentiation in transfected PC12 cells. J Biol Chem 273: 24624–24632.
Woolverton WL (1987). Evaluation of the role of norepinephrine in the reinforcing effects of psychomotor stimulants in rhesus monkeys. Pharmacol Biochem Behav 26: 835–939.
Wurtman RJ (2002). Stress and the adrenocortical control of epinephrine synthesis. Metab Clin Exp 51: 11–14.
Yang X, Chiba S (2002). Effects of L-765,314, a selective and potent α1B-adrenoceptor antagonist, on periarterial nerve electrical stimulation-induced double-peaked constrictor responses in isolated dog splenic arteries. Jpn J Pharmacol 89: 429–432.
Yang X-M, Gorman AL, Dunn AJ (1990). The involvement of central noradrenergic systems and corticotropin-releasing factor in defensive-withdrawal in rats. J Pharmacol Exp Ther 255: 1064–1070.
Yang M, Reese J, Cotecchia S, Michel MC (1998). Murine alpha1-adrenoceptor subtypes. I. Radioligand binding studies. J Pharmacol Exp Ther 286: 841–847.
Yang MY, Verfurth F, Buscher R, Michel MC (1997). Is α1D-adrenoceptor protein detectable in rat tissues? Naunyn Schmiedebergs Arch Pharmacol 355: 438–446.
Yntema OP, Korf J (1987). Transient suppression by stress of haloperidol induced catalepsy by the activation of the adrenal medulla. Psychopharmacology 91: 131–134.
Zebrowska-Lupina I, Przegalinski E, Sloniec M, Kleinrok Z (1977). Clonidine-induced locomotor hyperactivity in rats: the role of central postsynaptic α-adrenoceptors. Naunyn Schmiedeberg's Arch Pharmacol 297: 227–231.
Zebrowska-Lupina I, Stelmasiak M, Porowska A, Pietrasiewicz T (1988). Immobilization stress modifies locomotor response to catecholamine receptor agonists in rats. Pol J Pharmacol Pharm 40: 441–450.
Zhang Y, Itteera A, Quartermain D, Stone EA (2002). Effects of stress, corticosterone and LPS on alpha 1-adrenergically induced motor hyperactivity. Abstr Soc Neurosci 26, Prog #45.4.
Zilles K, Qu M, Schleicher A (1993). Regional distribution and heterogeneity of alpha-adrenoceptors in the rat and human central nervous system. J Hirnforschung 34: 123–132.
Zuscik MJ, Sands S, Ross SA, Waugh DJJ, Gaivin RJ, Morilak D et al (2000). Overexpression of the α1B-adrenergic receptor causes apoptotic neurodegeneration: multiple system atrophy. Nat Med 6: 1388–1394.
Acknowledgements
We thank Dr Kenneth Minneman of Emory University for his helpful discussion of the experiments reviewed in this work. This work was supported by Grant MH45265 from the NIMH.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stone, E., Lin, Y., Rosengarten, H. et al. Emerging Evidence for a Central Epinephrine-Innervated α1-Adrenergic System that Regulates Behavioral Activation and is Impaired in Depression. Neuropsychopharmacol 28, 1387–1399 (2003). https://doi.org/10.1038/sj.npp.1300222
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300222
Keywords
This article is cited by
-
Central norepinephrine transmission is required for stress-induced repetitive behavior in two rodent models of obsessive-compulsive disorder
Psychopharmacology (2020)
-
Relationships Between Catecholamine Levels and Stress or Intelligence
Neurochemical Research (2019)
-
The development of depression-like behavior is consolidated by IL-6-induced activation of locus coeruleus neurons and IL-1β-induced elevated leptin levels in mice
Psychopharmacology (2016)
-
A Mechanism for Nerve Cell Excitation by Norepinephrine via Alpha-1 Adrenoceptors: Inhibition of Potassium M-Current
Cellular and Molecular Neurobiology (2013)
-
Epinephrine: A Short- and Long-Term Regulator of Stress and Development of Illness
Cellular and Molecular Neurobiology (2012)
