
Abstract
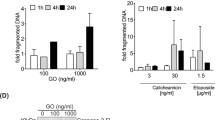
Calicheamicin ϑII is a member of the enediyne class of antitumor antibiotics that bind to DNA and induce apoptosis. These compounds differ, however, from conventional anticancer drugs as they bind in a sequence-specific manner noncovalently to DNA and cause sequence-selective oxidation of deoxyriboses and bending of the DNA helix. Calicheamicin is clinically employed as immunoconjugate to antibodies directed against, for example, CD33 in the case of gemtuzumab ozogamicin. Here, we show by the use of the unconjugated drug that calicheamicin-induced apoptosis is independent from death-receptor/FADD-mediated signals. Moreover, calicheamicin triggers apoptosis in a p53-independent manner as shown by the use of p53 knockout cells. Cell death proceeds via activation of mitochondrial permeability transition, cytochrome c release and activation of caspase-9 and -3. The overexpression of Bcl-xL or Bcl-2 strongly inhibited calicheamicin-induced apoptosis. Knockout of Bax abrogated cell death after calicheamicin treatment. Thus, the activation of mitochondria and execution of cell death occur through a fully Bax-dependent mechanism. Interestingly, caspase inhibition by the pancaspase-inhibitor zVAD-fmk interfered with mitochondrial activation by calicheamicin. This places caspase activation upstream of the mitochondria and indicates that calicheamicin-triggered apoptosis is enhanced through death receptor-independent activation of the caspase cascade, that is, an amplification loop that is required for full activation of the mitochondrial pathway.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout











Similar content being viewed by others

References
Ameisen JC . (2002). Cell Death Differ., 9, 367–393.
Belka C and Budach W . (2002). Int. J. Radiat. Biol., 78, 643–658.
Belka C, Rudner J, Wesselborg S, Stepczynska A, Marini P, Lepple-Wienhues A, Faltin H, Bamberg M, Budach W and Schulze-Osthoff K . (2000). Oncogene, 19, 1181–1190.
Belka C, Schmid B, Marini P, Durand E, Rudner J, Faltin H, Bamberg M, Schulze-Osthoff K and Budach W . (2001). Oncogene, 20, 2190–2196.
Bosanquet AG, Sturm I, Wieder T, Essmann F, Bosanquet MI, Head DJ, Dörken B and Daniel PT . (2002). Leukemia, 16, 1035–1044.
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B . (1998). Science, 282, 1497–1501.
Cohen GM . (1997). Biochem. J., 326, 1–16.
Cortazzo M and Schor NF . (1996). Cancer Res., 56, 1199–1203.
Daniel PT . (2000). Leukemia, 14, 2035–2044.
Daniel PT, Schulze-Osthoff K, Belka C and Güner D . (2003). Essays Biochem., 39, (in press).
Daniel PT, Sturm I, Ritschel S, Friedrich K, Dörken B, Bendzko P and Hillebrand T . (1999). Anal. Biochem., 266, 110–115.
Daniel PT, Sturm I, Wieder T and Schulze-Osthoff K . (2001). Leukemia, 15, 1022–1032.
de Vetten MP, Jansen JH, van der Reijden BA, Berger MS, Zijlmans JM and Lowenberg B . (2000). Br. J. Haematol., 111, 277–279.
Dhein J, Daniel PT, Trauth BC, Oehm A, Moller P and Krammer PH . (1992). J. Immunol., 149, 3166–3173.
Distelhorst CW . (2002). Cell Death Differ., 9, 6–19.
Engels IH, Stepczynska A, Stroh C, Lauber K, Berg C, Schwenzer R, Wajant H, Janicke RU, Porter AG, Belka C, Gregor M, Schulze-Osthoff K and Wesselborg S . (2000). Oncogene, 19, 4563–4573.
Essmann F, Wieder T, Otto A, Muller EC, Dörken B and Daniel PT . (2000). Biochem. J., 346, 777–783.
Fadok VA, Xue D and Henson P . (2001). Cell Death Differ., 8, 582–587.
Friedrich K, Wieder T, Von Haefen C, Radetzki S, Janicke R, Schulze-Osthoff K, Dörken B and Daniel PT . (2001). Oncogene, 20, 2749–2760.
Fulda S, Meyer E, Friesen C, Susin SA, Kroemer G and Debatin KM . (2001). Oncogene, 20, 1063–1075.
Gillissen B, Essmann F, Graupner V, Stärck L, Radetzki S, Dörken B, Schulze-Osthoff K and Daniel PT . (2003). EMBO J., 22, 3580–3590.
Güner D, Sturm I, Hemmati PG, Hermann S, Hauptmann S, Wurm R, Budach V, Dörken B, Lorenz M and Daniel PT . (2003). Int. J. Cancer, 103, 445–454.
Hemmati PG, Gillissen B, von Haefen C, Wendt J, Stärck L, Güner D, Dörken B and Daniel PT . (2002). Oncogene, 21, 3149–3161.
Hiatt A, Merlock R, Mauch S and Wrasidlo W . (1994). Bioorg. Med. Chem., 2, 315–322.
Koonin EV and Aravind L . (2002). Cell Death Differ., 9, 394–404.
Kroemer G and Reed JC . (2000). Nat. Med., 6, 513–519.
Lassus P, Opitz-Araya X and Lazebnik Y . (2002). Science, 297, 1352–1354.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES and Wang X . (1997). Cell, 91, 479–489.
Linenberger ML, Hong T, Flowers D, Sievers EL, Gooley TA, Bennett JM, Berger MS, Leopold LH, Appelbaum FR and Bernstein ID . (2001). Blood, 98, 988–994.
Liu X, Kim CN, Yang J, Jemmerson R and Wang X . (1996). Cell, 86, 147–157.
Maiese WM, Lechevalier MP, Lechevalier HA, Korshalla J, Kuck N, Fantini A, Wildey MJ, Thomas J and Greenstein M . (1989). J. Antibiot., 42, 558–563.
Marsden VS, O’Connor L, O’Reilly LA, Silke J, Metcalf D, Ekert PG, Huang DC, Cecconi F, Kuida K, Tomaselli KJ, Roy S, Nicholson DW, Vaux DL, Bouillet P, Adams JM and Strasser A . (2002). Nature, 419, 634–637.
Martinou JC and Green DR . (2001). Nat. Rev. Mol. Cell. Biol., 2, 63–67.
Mendelsohn AR, Hamer JD, Wang ZB and Brent R . (2002). Proc. Natl. Acad. Sci. USA, 99, 6871–6876.
Mrozek A, Petrowsky H, Sturm I, Krauss J, Hermann S, Hauptmann S, Lorenz M and Daniel P . (2003). Cell Death Differ, 10, 461–467.
Nabhan C and Tallman MS . (2002). Clin. Lymphoma, 2 (Suppl. 1), S19–S23.
Nicolaou KC, Li T, Nakada M, Hummel W, Hiatt A and Wrasidlo W . (1994a). Angew. Chem., 33, 183–186.
Nicolaou KC, Pitsinos EN, Theodorakis EA, Saimoto H and Wrasidlo W . (1994b). Chem. Biol., 1, 57–66.
Nicolaou KC, Stabila P, Esmaeli-Azad B, Wrasidlo W and Hiatt A . (1993). Proc. Natl. Acad. Sci. USA, 90, 3142–3146.
Prokop A, Wieder T, Sturm I, Essmann F, Seeger K, Wuchter C, Ludwig W-D, Henze G, Dörken B and Daniel PT . (2000). Leukemia, 14, 1606–1613.
Radetzki S, Kohne CH, von Haefen C, Gillissen B, Sturm I, Dörken B and Daniel PT . (2002). Oncogene, 21, 227–238.
Raisova M, Bektas M, Wieder T, Daniel P, Eberle J, Orfanos CE and Geilen CC . (2000). FEBS Lett., 473, 27–32.
Raisova M, Hossini A, Eberle J, Riebeling C, Wieder T, Sturm I, Daniel PT, Orfanos CE and Geilen CC . (2001). J. Invest. Dermatol., 177, 333–340.
Robertson JD, Enoksson M, Suomela M, Zhivotovsky B and Orrenius S . (2002). J. Biol. Chem., 277, 29803–29809.
Roucou X and Martinou JC . (2001). Cell Death Differ., 8, 875–877.
Rudner J, Jendrossek V and Belka C . (2002). Apoptosis, 7, 441–447.
Rudner J, Lepple-Wienhues A, Budach W, Berschauer J, Friedrich B, Wesselborg S, Schulze-Osthoff K and Belka C . (2001). J. Cell Sci., 114, 4161–4172.
Salvesen GS and Dixit VM . (1997). Cell, 91, 443–446.
Salzberg AA and Dedon PC . (2000). Biochemistry, 39, 7605–7612.
Schlegel RA and Williamson P . (2001). Cell Death Differ., 8, 551–563.
Schor NF, Tyurina YY, Fabisiak JP, Tyurin VA, Lazo JS and Kagan VE . (1999). Brain Res., 831, 125–130.
Sievers EL, Appelbaum FR, Spielberger RT, Forman SJ, Flowers D, Smith FO, Shannon-Dorcy K, Berger MS and Bernstein ID . (1999). Blood, 93, 3678–3684.
Slee EA, Keogh SA and Martin SJ . (2000). Cell Death Differ., 7, 556–565.
Smaili SS, Hsu YT, Sanders KM, Russell JT and Youle RJ . (2001). Cell Death Differ., 8, 909–920.
Sturm I, Bosanquet A, Hermann S, Güner D, Dörken B and Daniel P . (2003). Cell Death Differ, 10, 477–484.
Sturm I, Petrowsky H, Volz R, Lorenz M, Radetzki S, Hillebrand T, Wolff G, Hauptmann S, Dörken B and Daniel PT . (2001). J. Clin. Oncol., 19, 2272–2281.
Suzuki A, Tsutomi Y, Miura M and Akahane K . (1999). Oncogene, 18, 1239–1244.
Thorson JS, Sievers EL, Ahlert J, Shepard E, Whitwam RE, Onwueme KC and Ruppen M . (2000). Curr. Pharm. Des., 6, 1841–1879.
Tsujimoto Y and Shimizu S . (2000). Cell Death Differ., 7, 1174–1181.
Utz PJ and Anderson P . (2000). Cell Death Differ., 7, 589–602.
van der Velden VH, te Marvelde JG, Hoogeveen PG, Bernstein ID, Houtsmuller AB, Berger MS and van Dongen JJ . (2001). Blood, 97, 3197–3204.
van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ and Vandenabeele P . (2002). Cell Death Differ., 9, 1031–1042.
Vermes I, Haanen C, Steffens-Nakken H and Reutelingsperger C . (1995). J. Immunol. Methods, 184, 39–51.
von Ahsen O, Waterhouse NJ, Kuwana T, Newmeyer DD and Green DR . (2000). Cell Death Differ., 7, 1192–1199.
von Haefen C, Wieder T, Essmann F, Schulze-Osthoff K, Dörken B and Daniel PT . (2003). Oncogene, 22, 2236–2247.
von Haefen C, Wieder T, Gillissen B, Stärck L, Graupner V, Dörken B and Daniel PT . (2002). Oncogene, 21, 4009–4019.
Wieder T, Essmann F, Prokop A, Schmelz K, Schulze-Osthoff K, Beyaert R, Dörken B and Daniel PT . (2001a). Blood, 97, 1378–1387.
Wieder T, Geilen CC, Kolter T, Sadeghlar F, Sandhoff K, Brossmer R, Ihrig P, Perry D, Orfanos CE and Hannun YA . (1997). FEBS Lett., 411, 260–264.
Wieder T, Orfanos CE and Geilen CC . (1998). J. Biol. Chem., 273, 11025–11031.
Wieder T, Prokop A, Bagci B, Essmann F, Bernicke D, Schulze-Osthoff K, Dörken B, Schmalz HG, Daniel PT and Henze G . (2001b). Leukemia, 15, 1735–1742.
Zhang L, Yu J, Park BH, Kinzler KW and Vogelstein B . (2000). Science, 290, 989–992.
Acknowledgements
This work was supported by grants from the Deutsche José Carreras Leukämie-Stiftung, the Verein zur Förderung der Tagesklinik and the Deutsche Krebshilfe. We thank B Vogelstein (Johns Hopkins University School of Medicine, Baltimore, MD, USA) for kindly providing HCT116 wild-type, p53 −/− and Bax −/− cells and the congeneic controls, S Fulda (University of Ulm, Germany) for kindly providing BJAB cells overexpressing Bcl-xL and K Schulze-Osthoff (University of Düsseldorf, Germany) for kindly providing Jurkat cells overexpressing Bcl-2. The excellent technical assistance of A Richter is gratefully acknowledged.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Prokop, A., Wrasidlo, W., Lode, H. et al. Induction of apoptosis by enediyne antibiotic calicheamicin ϑII proceeds through a caspase-mediated mitochondrial amplification loop in an entirely Bax-dependent manner. Oncogene 22, 9107–9120 (2003). https://doi.org/10.1038/sj.onc.1207196
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1207196
Keywords
This article is cited by
-
Inotuzumab ozogamicin as single agent in pediatric patients with relapsed and refractory acute lymphoblastic leukemia: results from a phase II trial
Leukemia (2022)
-
Fate of Antibody-Drug Conjugates in Cancer Cells
Journal of Experimental & Clinical Cancer Research (2018)
-
Cytokine-induced senescence for cancer surveillance
Cancer and Metastasis Reviews (2017)
-
Current ADC Linker Chemistry
Pharmaceutical Research (2015)
-
Iron containing anti-tumoral agents: unexpected apoptosis-inducing activity of a ferrocene amino acid derivative
Journal of Cancer Research and Clinical Oncology (2011)
