
Abstract
Execution of the intrinsic apoptotic pathway is controlled by the BCL-2 proteins at the level of the mitochondrial outer membrane (MOM). This family of proteins consists of prosurvival (e.g., BCL-2, MCL-1) and proapoptotic (e.g., BIM, BAD, HRK) members, the functional balance of which dictates the activation of BAX and BAK. Once activated, BAX/BAK form pores in the MOM, resulting in cytochrome c release from the mitochondrial intermembrane space, leading to apoptosome formation, caspase activation, and cleavage of intracellular targets. This pathway is induced by cellular stress including DNA damage, cytokine and growth factor withdrawal, and chemotherapy/drug treatment. A well-documented defense of leukemia cells is to shift the balance of the BCL-2 family in favor of the prosurvival proteins to protect against such intra- and extracellular stimuli. Small molecule inhibitors targeting the prosurvival proteins, named ‘BH3 mimetics’, have come to the fore in recent years to treat hematological malignancies, both as single agents and in combination with standard-of-care therapies. The most significant example of these is the BCL-2-specific inhibitor venetoclax, given in combination with standard-of-care therapies with great success in AML in clinical trials. As the number and variety of available BH3 mimetics increases, and investigations into applying these novel inhibitors to treat myeloid leukemias continue apace the need to evaluate where we currently stand in this rapidly expanding field is clear.
Similar content being viewed by others


Facts
-
Dysregulation of prosurvival BCL-2 proteins is highly implicated in the oncogenesis, progression, and therapy-resistance of myeloid leukemias.
-
BH3 mimetics inhibit prosurvival BCL-2 proteins and re-balance the apoptotic pathway.
-
The BCL-2-specific BH3 mimetic venetoclax has had significant clinical success in acute myeloid leukemia in combination with standard therapy.
-
Various BH3 mimetics in combination with standard-of-care therapies are currently under investigation in myeloid leukemias.
Open Questions
-
Could BH3 mimetics be useful in the treatment of chronic myeloid leukemia, either alone or in combination with tyrosine kinase inhibitors?
-
Will the recent clinical success of the BCL-2 inhibitor venetoclax in combination with cytarabine or hypomethylating agents in acute myeloid leukemia encourage further combinatorial studies of venetoclax with other standard-of-care therapies in this disease?
-
Is there scope for using MCL-1 and BCL-xL inhibitors in myeloid leukemias in a clinical setting?
Introduction
Apoptosis is the best-described form of programmed cell death, discrete from other forms of cell death such as autophagy, necroptosis, ferroptosis, and pyroptosis1,2. An apoptotic cell displays morphological changes including nucleus shrinkage and membrane blebbing. Apoptotic cells undergo DNA degradation, cleavage of intracellular structures, and loss of mitochondrial function1. The term ‘apoptosis’ refers to two pathways distinct in initiation. The extrinsic apoptotic pathway is triggered via death receptor binding at the cellular membrane3, while the intrinsic, or ‘mitochondrial’, the pathway is regulated by B-cell leukemia/lymphoma-2 (BCL-2) family of proteins4,5. The two pathways crosstalk at the level of truncated BH3-interacting domain death agonist (tBID) activation, which occurs concurrently with the instigation of a caspase cascade in the context of the extrinsic pathway, and prior to activation of the multidomain proapoptotic effector proteins in the case of the intrinsic pathway.
During neoplastic transformation cells face numerous signals, including DNA damage, which would initiate apoptosis in healthy cells; however malignant cells hijack the apoptotic machinery to evade cell death6. One extensively studied example is an over-reliance on the BCL-2 family7. BCL-2 was first described in relation to survival from cell death due to its role as a driver of follicular lymphoma (FL)8. Other prosurvival members of the BCL-2 family have been implicated to varying degrees in the pathogenesis of other hematological malignancies including acute myeloid leukemia (AML)9,10, chronic myeloid leukemia (CML)11,12,13,14,15, multiple myeloma16, diffuse large B-cell lymphoma17, and acute lymphoblastic leukemia (ALL)18.
In recent years, the implications of BCL-2 family dependence in hematological malignancies has resulted in widespread and sustained effort to investigate whether this can be exploited to selectively eliminate cancerous cells.
The intrinsic apoptotic pathway
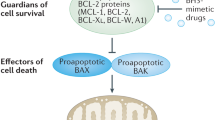
Within the intrinsic apoptotic pathway, the decision to commit to cell death occurs at the mitochondrial outer membrane (MOM) and is dictated by a balance between opposing factions within the BCL-2 family19. The family can be divided primarily by function (prosurvival or proapoptotic), and latterly by structure (Fig. 1). BCL-2, along with myeloid cell leukemia-1 (MCL-1), B-cell lymphoma-extra large (BCL-xL; BCL2L1), B-cell lymphoma-w (BCL-w), and BCL-2-related gene expressed in fetal liver-1 (Bfl-1; A1), are able to inhibit apoptosis and contain three to four regions of conserved homology termed BCL-2 homology (BH) domains 1-4. Within the proapoptotic group of proteins there are two subgroups: (1) the multidomain proteins BCL-2-associated X protein (BAX) and BCL-2 homologous antagonist killer (BAK), and; (2) the BH3-only proteins, including BCL-2-interacting mediator of cell death (BIM), a p53-upregulated modulator of apoptosis (PUMA; BBC3), BCL-2 associated death promoter (BAD), NOXA (phorbol-12 myristate-13-acetate-induced protein 1; PMAIP1), BH3-interacting domain (BID), BCL-2-interacting killer (BIK), BCL-2-modifying factor (BMF) and Harakiri (HRK).

Figure not to scale. TM transmembrane domain.
When activated, BAX and BAK oligomerize to form toroidal structures within the membrane20. The result is MOM permeabilisation (MOMP), the efflux of proteins from the mitochondrial intermembrane space and subsequent loss of membrane potential. Among the proteins released from the mitochondria through the BAX/BAK pores is cytochrome c, which combines with apoptotic protease-activating factor 1 (APAF1) and caspase-9 to form the apoptosome, a multi-protein complex that activates the effector caspases-3, -6 and -721.
The mechanism of BAX and BAK activation may occur via the direct activation and/or the indirect activation model (Fig. 2)22. The direct activation model suggests that BAX and BAK exist in an inactive conformation until activated by a subset of BH3-only proteins termed ‘direct activators’, including BIM, PUMA, and BID. These direct activators are sequestered by the prosurvival proteins and released upon inhibition of the latter by further ‘sensitiser’ BH3-only proteins (such as NOXA, HRK, and BAD). Conversely, the indirect activation model postulates that BAX/BAK are constitutively active and are inhibited by the prosurvival BCL-2 proteins; in response to a death stimulus, BH3-only proteins in turn inhibit the prosurvival proteins, thereby lifting the inhibition of BAX/BAK. In both models, the inhibition of the prosurvival proteins and subsequent release of either direct activator BH3-only proteins or BAX/BAK is the initiating step in triggering apoptosis23.

BAX/BAK activation occurs through either the direct activation model, indirect activation model or a balance of both.
BH3-only proteins display differing binding affinities to the prosurvival proteins (e.g., NOXA binds with high specificity to MCL-1, while HRK binds exclusively with BCL-xL)24. Along with the evidence that malignant cells can evade apoptosis through over-reliance on the prosurvival BCL-2 proteins, this has led to the development of highly specific small molecule inhibitors of the prosurvival proteins. The inhibitors are rationally designed to mimic the BH3-only protein known to inhibit the prosurvival protein of interest and, as such, are termed ‘BH3 mimetics’25.
In this review, we focus on the use of BH3 mimetics within the myeloid leukemias, specifically CML and AML. We highlight the dependencies on these proteins, the compounds developed to take advantage of these discoveries and investigations conducted which combine BH3 mimetics with standard-of-care therapies, concluding with future directions for the field.
Dysregulation of the intrinsic apoptotic pathway in CML
CML is typified by the Philadelphia chromosome, a (t(9;22)(q34;q11)) chromosomal translocation arising in a hematopoietic stem cell (HSC), leading to the expression of the fusion oncoprotein BCR-ABL26,27. This constitutively active tyrosine kinase sits at the epicenter of a complex signaling network that contributes to the malignant transformation of HSC into leukemic stem cells (LSC) which overpopulate the hematopoietic system with a myeloid bias28.
The current standard-of-care treatment for chronic phase (CP) CML is BCR-ABL-specific tyrosine kinase inhibitors (TKIs) such as imatinib (Gleevec®)29. For patients whose cancer becomes TKI-refractory, the disease may progress to blast phase (BP), with a 6 to 11-month survival rate and few treatment options available30. The BCL-2 family of proteins has been implicated in both CML development and progression; therefore, targeting the intrinsic apoptotic pathway may be a viable therapeutic option (Fig. 3).

Targeting of these protein results in a shift away from apoptosisin CML cells. Black arrows indicate BCR-ABL-mediated up- or downregulation.
BIM in CML
Evasion of apoptosis in response to cytokine withdrawal is one of the most consistently observed effects of BCR-ABL; this withdrawal results in BIM upregulation in normal hematopoietic progenitors, but the effect is abolished in BCR-ABL-transformed cells31. BCR-ABL downregulates BIM transcription and labels the BIM protein for degradation through mitogen-activated protein kinase (MAPK) phosphorylation, with BIM levels restored by inhibiting BCR-ABL with TKIs, such as imatinib. Silencing of BIM effectively rescues CML cells from apoptosis caused by imatinib31,32. BCR-ABL therefore supports CML cell survival, at least in part, through the downregulation of BIM.
Prosurvival BCL-2 proteins
MCL-1 mRNA and MCL-1 protein are expressed constitutively in a BCR-ABL-dependent manner in CML regardless of disease stage12. Upregulation of BCL-xL has been observed in BCR-ABL-transformed HL-60 and BaF3 cells, while inhibition of the Akt/Protein kinase B pathway was found to reverse the upregulation of BCL-xL in the latter33,34. Investigations using apoptosis-resistant BCR-ABL+ mice suggest BCL-2 mutations in myeloid progenitors may be critical in the transition of BCR-ABL+ leukemias to advanced stage disease13. Further, inhibiting both BCL-2 and BCR-ABL is sufficient to induce apoptosis in CML stem cells in a murine CML model and TKI-resistant BP-CML patient samples35.
Role of BAX
The serine/threonine-specific protein kinase Akt/Protein kinase B, a downstream target of BCR-ABL, is constitutively active in CP-CML and BP-CML cells; Akt inhibits a conformational change in BAX required for translocation to the mitochondrial membrane, thus hindering MOMP in response to cellular stress36,37. In CML cells expressing high levels of BCR-ABL, this movement of BAX is prevented38.
The microRNA miR-29b, able to increase the expression of BAX, is inhibited by BCR-ABL and is downregulated in BP-CML39,40. Overexpression of miR-29b in the CML cell line K562 has been shown to halt proliferation and induce apoptosis, indicating an important role for this miR in regulating cell death39.
Thus, CML cells can hijack BAX both at the translational and conformational levels, thereby decreasing sensitivity to cytotoxic stimuli and a further balance shift of the BCL-2 family proteins in favor of cell survival.
Dysregulation of the intrinsic apoptotic pathway—AML
AML is the most common myeloid malignancy in adults, with an incidence rate of 3–5 cases per 100,000 per year and a median age of 68 years at diagnosis. AML covers a genetically heterogenous group of disorders of myelopoiesis with immature myeloid blasts in the bone marrow, blood, and extramedullary tissues. These blast cells out-compete normal hematopoiesis leading to the disease phenotype of fever, infection, anemia, bruising, and bleeding41.
Classification of AML is based on the World Health Organization and European Leukemia Network criteria, which rely on morphology, immunophenotyping, and the detection of underlying genetic lesions including both recurrent cytogenetic and molecular abnormalities42. Conventional karyotyping is the mainstay of risk stratification in AML and is complemented by fluorescence in situ hybridization analysis and RT-PCR for the targeted detection of specific recurrent genetic abnormalities43.
Next-generation sequencing can further stratify AML based on the presence or absence of cooperating mutations involved in driving the disease, encompassing epigenetic regulators, cell signaling and proliferation pathways, master hematopoietic transcription factors, and tumor suppressors44. The cytogenetic and molecular abnormalities present at diagnosis influence prognosis and clinical management and are used to subtype patients appropriately into favorable, intermediate, and adverse prognostic categories41,42.
This complex genomic landscape, combined with other co-morbidities and age at onset, makes treatment and management of AML patients challenging and, increasingly, an individualized approach is required. The therapeutic pathway taken will depend not only on the underlying genomic lesions, but also the age and fitness of the patient. Younger and fit older patients will receive high-intensity induction chemotherapy, followed by either consolidation chemotherapy or a stem cell transplant, dependent on response to therapy and genetic lesions present at diagnosis. Until recently, patients deemed unfit to tolerate intensive chemotherapy would receive either low dose cytarabine (LDAC), hypomethylating agents (HMA), or palliative treatment such as hydroxyurea in addition to supportive care41. LDAC and HMA may achieve remissions in a minority of patients, but are not curative and almost all patients will relapse. Small molecule inhibitors may also be included for patients with specific genetic lesions, e.g. midostaurin for patients with a fms-like tyrosine kinase 3 (FLT3) mutation45.
Role of BCL-2 family in AML
The journey from the identification of BCL-2 dependence in AML to the successful development and clinical application of the BCL-2-specific inhibitor venetoclax is a triumph of modern cancer therapy. BCL-2 was found to be expressed in AML CD34+ progenitor cells and promyelocytes while this expression was absent in their heathy counterparts, and evidence was presented that induction chemotherapy resulted in selection for leukemic CD34+ cells expressing high levels of BCL-246. Later, it was shown that BCL-2 is essential for the maintenance of cancer cells in a murine model of leukemia, in the first example of the functional removal of a BCL-2 family prosurvival protein resulting in cancer regression47.
BCL-2 expression has also been shown to be significantly upregulated in newly diagnosed AML patients (range of 34–87%) and relapsed AML patients10,48,49,50. Patients with elevated BCL-2 tend to present with higher percentage of peripheral blasts48, with over-expression also correlating with CD34 and CD117 positivity and poorer response to chemotherapy10,49,50, suggesting a more primitive phenotype.
An early investigation saw the application of a cell-permeable BCL-2 binding peptide, based on the structure of BAD, in HL-60 cells in vitro and human myeloid leukemia cells in a murine model, resulting in leukemic cell death51. This was followed swiftly by the description of HA14-1, a small molecule compound able to bind to the BCL-2 surface pocket and capable of inducing caspase-dependent apoptosis in HL-60 cells52.
Other BCL-2 family proteins, including BCL-xL and MCL-1 have been implicated in the pathogenesis of AML53,54,55. BCL-xL and BAD, along with BCL-2, are upregulated in the majority of AML stem/progenitor cell populations, compared to normal hematopoietic stem/progenitor cells (HSPCs), with induction chemotherapy resulting in a further upregulation of BCL-2 and BCL-xL46,54. MCL-1 is consistently high in the majority of newly diagnosed AML patients and has been associated with relapse56,57. MCL-1 is also linked to stem cell survival, especially in FLT3-internal tandem duplication (FLT3-ITD) AML stem cells58.
A critical role of MCL-1 in cell survival was demonstrated in an elegant study using bone marrow HSCs/HSPCs transformed with the oncogenes mixed-lineage leukemia (MLL)-eleven nineteen leukemia (MLL-ENL) and MLL-ALL1-fused gene from chromosome 9 (MLL-AF9), and corresponding AML mouse models. Depletion of Mcl-1 led to the death of cells in vitro and reduced disease burden in AML-afflicted mice, with cell death being rescued by overexpressing Bcl-2 or Mcl-157.
Due to the heterogeneity of AML, studies indicate that cells may be ‘addicted’ to BCL-2, MCL-1, or both depending on the genomic landscape of the patient at diagnosis59. If BH3 mimetics are to be used successfully clinically in the management of AML, patient-specific prediction of BCL-2 family dependency, potentially by BH3 profiling may well be essential60,61.
The rise of BH3 mimetics
The developmental journey of BH3 mimetics to clinical use has been extensively covered, including the excellent reviews by Lessene et al.62 and Leverson et al.63. One of the first BH3 mimetics developed, following HA14-1, was ABT-737, a small molecule with high binding affinity to BCL-2, BCL-xL, and BCL-w16,64. ABT-737 represents the first example of an anti-cancer drug designed specifically to target a protein-protein interaction, and was identified through the structure-activity relationships (SAR) by nuclear magnetic resonance (NMR) method and site-directed parallel synthesis, a triumph of modern, rational cancer therapy design64.
The major limitation of ABT-737 was the lack of oral bioavailability, prompting the development of ABT-263 (navitoclax)65. Navitoclax is a dual inhibitor of BCL-2/BCL-xL, and its application as a monotherapy in relapsed/refractory (R/R) CLL was promising, with a 35% partial response rate, though 28% of patients experienced grade 3/4 thrombocytopenia due to the requirement of BCL-xL in the development of platelets66,67. The serious adverse effects associated with BCL-xL inhibition in vivo was addressed with the development of ABT-199 (venetoclax, Venclexta®), a highly specific BCL-2 inhibitor that induced less thrombocytopenia68.
Venetoclax was first described in 201368, and since has been approved in the US for the treatment of CLL with 17p deletion (2016), in combination with rituximab (Rituxan®) for previously untreated CLL (2018), newly diagnosed AML in combination with HMA or LDAC where intensive induction chemotherapy is not possible (accelerated approval in 2018, full approval in 2020) and in combination with non-chemotherapeutics for previously untreated CLL (2019). The path to these approvals in AML will be addressed further in this review.
Of interest in the context of the current COVID-19 pandemic, NHS England granted temporary emergency approval of venetoclax in specific AML patient groups. Venetoclax treatment can be delivered on an outpatient basis, allowing for reduced attendance at the clinic for the duration of the pandemic until regular treatment can be resumed.
Resistance to venetoclax can occur through upregulation of other BCL-2 prosurvival proteins, and subsequent targeting of these proteins with alternative BH3 mimetics or inhibiting upstream regulatory pathways is often effective in overcoming resistance69,70,71. To this end, and especially when targeting cancers in which BCL-2 is not the primary prosurvival BCL-2 protein, other BH3 mimetics may come to the fore of clinical studies in the future (Table 1).
BH3 mimetics with standard-of-care therapies
Two methods of administering BH3 mimetics deserve consideration: co-treatment and the one-two punch method (Fig. 4). In the first, a BH3 mimetic is chosen based on the known BCL-2 dependency of the cancer cells to prohibit this defense against the chemotherapy of choice. The second method involves inducing a targetable change in the cancer cells. Treatment with the chemotherapy is used to induce cell death signaling in the cells, thereby making the cells more reliant on one or more of the BCL-2 family proteins; the cells can then be targeted with a selected BH3 mimetic. To be most effective, this may require several rounds of sequential treatment with chemotherapy, alternating with a BH3 mimetic. In both cases, dependency can be measured through ex vivo mimetic treatment or methods such as BH3 profiling72. This approach may be particularly valuable in diseases such as AML where complex genetic landscapes make it more challenging to predict individual patient response to treatment.

Proposed methods for combination treatments with BH3 mimetics include co-treatment with a chemotherapy or inducing an increase in dependency on a BCL-2 prosurvival protein by treatment with chemotherapy which can then be targeted with a BH3 mimetic (one-two punch method).
Although a vast array of highly specific BH3 mimetics targeting different members of the BCL-2 family are available to researchers at the bench, venetoclax is the only one in common use clinically. This is due to the adverse effects of targeting BCL-xL and MCL-1, namely thrombocytopenia66,73 and potentially cardiac toxicity74,75, respectively. If BH3 mimetics targeting other family members are to be used clinically in the future, fine-tuning to improve tolerability will be required. Drug dosages that induce cancer cell death must be lower than those which damage healthy tissue; it is here that the one-two punch method could be utilized, to heighten the sensitivity of the cancer cells to the mimetic, and to allow recovery of normal tissue in between rounds of mimetic treatment.
Chronic myeloid leukemia
One of the biggest challenges in treating CP-CML is TKI-resistance. Among other pathways, overexpression of the BCL-2 prosurvival proteins and low levels of the proapoptotic BIM protein have been linked to TKI-resistance, leading to investigations into combining TKIs with BH3 mimetics. The persistence of CML stem cells also represents a barrier to the successful elimination of the disease;28 these LSCs are resistant to TKI treatment, with alternative methods for eradicating this population therefore required76,77.
Pre-clinical combinations of TKIs with BH3 mimetics in CML
In terms of circumventing TKI-resistance via BCL-2 family imbalance mechanisms, co-treatment using TKI with a BH3 mimetic has shown efficacy. ABT-737 re-sensitized the CML cell line K562 to imatinib-induced cell killing in cells with imatinib-resistance mediated by BIM knockdown or BCL-2 overexpression32. This effect was also observed in Bim−/−Bad−/− BCR-ABL-transformed murine fetal liver-derived myeloid progenitor cells. These findings demonstrate that imatinib-resistance resulting from alterations in the BCL-2 family can be overcome through co-treatment with a BH3 mimetic
Analysis of CP-CML East Asian patients found a BIM deletion polymorphism, resulting in expression of BIM lacking the BH3 domain, and was linked to TKI-resistance78. Crucially, although TKI-resistance is usually associated with BCR-ABL kinase domain mutations79, it was found that patients with the polymorphism were less likely to have a BCR-ABL kinase domain mutation, suggesting an almost mutually exclusive mechanism of TKI-resistance and that treating these patients with further TKIs may be of little advantage. Co-treatment with ABT-737, however, restored imatinib-induced cell death in BIM-mutated CML cell lines and patient samples with the polymorphism.
More recently, BH3 mimetics in combination with TKIs have been used to target the CML progenitor compartment, with notable success in a number of CML disease models35,80,81,82. These include reducing the colony-forming capacity of CP-CML progenitors (CD34+ CP-CML cells, venetoclax with imatinib)80, reducing leukemic burden and long-term engraftment potential and increasing overall survival in CML murine models (Bcr-Abl1+ Tet-off Lin-Sca-1+cKit+ cells, venetoclax with nilotinib)35, and increasing apoptosis in BP-CML progenitors (CD34+CD38− BP-CML cells, venetoclax with nilotinib)35, (CD34+ BP-CML cells, ABT-737 with imatinib)81 and CP-CML progenitors (CD34+CD38- CP-CML cells, ABT-737 with imatinib/nilotinib)82.
In summary, for TKI-resistance mediated by the proapoptotic or prosurvival proteins, BH3 mimetics appear effective in redressing the balance, and re-sensitizing CML cells to TKIs.
However, due to the effectiveness of TKIs alone, there is very little clinical trial activity investigating combinations of BH3 mimetics with TKIs. One phase 2 clinical trial is currently recruiting, combining venetoclax with the TKI dasatinib in early CP-CML, with the primary endpoint of assessing the proportion of patients achieving major molecular response after 12 months of therapy (NCT02689440)83. To date, no results are available for this trial. A second phase 2 study combining decitabine, ponatinib and venetoclax in blast phase CML is also underway, with a primary endpoint of overall response rate (NCT04188405)84. To date, there are no clinical trials in CML of MCL-1 or BCL-xL inhibitors.
Acute myeloid leukemia
The success story of venetoclax in AML is one that cannot be understated, especially for the exceptionally short timeframe from the first description of venetoclax in 201368 to full approval by the FDA for venetoclax plus HMA or LDAC in older, unfit AML patients in 2020. This speaks to the substantial and convincing work into the BCL-2 family in AML, through pre-clinical studies of venetoclax alone and in combination with other therapies, to the large international trials that resulted in directly improving patient care. Figure 5 illustrates this remarkable path.

Extensive experimental data identifying the prosurvival BCL-2 family members as potential therapeutic targets, focused preclinical work combining BH3 mimetics with standard-of-care therapies in AML, and highly promising, international clinical trials have led to FDA approval of venetoclax with HMA or LDAC in an impressively short timeframe.
Along with mounting evidence for the role of BCL-2 in AML cell survival, early preclinical studies into venetoclax as a monotherapy in AML cell lines, patient samples, and a murine xenograft model demonstrated on-target cell killing9, with particular sensitivity to venetoclax seen in AML cells harboring the MLL fusion genes and in acute promyelocytic leukemia (APL) cells85. Interestingly, venetoclax is especially potent in AML cells with isocitrate dehydrogenase 1 and 2 (IDH1/2) gene mutations; these proteins have been implicated in increasing BCL-2 dependence86. In a phase 2 clinical trial in relapsed/refractory AML, single-agent venetoclax had an overall response rate of 19%, while 33% (4 out of 12) of patients with IDH1/2 mutations demonstrated CR87,88.
There is also increasing use and success with the MCL-1 inhibitors AMG-176, AMG-397, and S64315 in pre-clinical models of AML, regardless of the presence of specific genetic lesions59,89.
Despite the limited clinical success of venetoclax as a monotherapy in AML, evidence supporting the combination of venetoclax with standard-of-care therapies in AML is encouraging. For example, treatment of AML patients with cytarabine and idarubicin has been shown to increase BCL-2 expression in the CD34+ compartment46 and high de novo expression of BCL-2 is correlated with poor response to treatment90,91, indicating scope for venetoclax combinations in AML.
Azacitidine with BH3 mimetics in AML
Pre-clinical synergistic cytotoxic effects were shown by several groups when combining ABT-737 or venetoclax with the HMA azacitidine in AML cell lines and primary patient samples in vitro92,93,94. RNA-interference screening identified the proapoptotic BCL-2 proteins as potential targets for enhancing the effects of azacitidine92, and later it was shown that ABT-737 was a more potent agent than venetoclax when used in combination with azacitidine, due to the variable expression of the prosurvival BCL-2 proteins between patients93.
These promising findings led to the development of clinical trials investigating a BCL-2-targeting BH3 mimetic in combination with azacitidine in myeloid leukemias. A phase 1b trial comparing the combination of venetoclax with azacitidine or decitabine in AML patients over 65 years of age with treatment-naïve AML, and who were ineligible for intensive chemotherapy, demonstrated an extremely promising 73% CR or CR with incomplete count recovery (CRi) for the cohort receiving HMA and 400 mg venetoclax95,96,97. These results led, in 2016, to the FDA granting venetoclax Breakthrough Therapy Designation in combination with HMA in older patients with treatment-naive AML.
The large VIALE-A phase 3 trial that followed combined azacitidine with 400 mg venetoclax and compared against an azacitidine plus placebo control group, enrolling 443 untreated AML patients who were either over the age of 75 or could not tolerate standard chemotherapy, or both98. At an interim analysis, overall survival (OS) and CR were increased from the control (OS: 9.6 months; CR: 28.3%) to the azacitidine plus venetoclax group (OS: 14.7 months; CR: 66.4%)99. By 2018, the FDA had granted accelerated approval for venetoclax in combination with HMA in patients who cannot receive induction chemotherapy, and full approval granted for this indication in 2020.
Cytarabine with BH3 mimetics in AML
Incorporation of cytarabine into the DNA of rapidly dividing cells induces cell cycle arrest in the S phase through inhibition of DNA synthesis100. Pre-clinical inhibition of BCL-2 by antisense oligonucleotides, obatoclax or venetoclax in combination with cytarabine has been shown to significantly enhance cell death in AML cell lines and patient samples71,101,102. Cytarabine-mediated reduction of MCL-1 expression may also contribute to the synergistic action of BCL-2 and/or BCL-xL inhibition in these cells71.
In phase 1b/2 clinical trials with patients over the age of 65, the combination of venetoclax with LDAC had a CR rate of 54% and OS of 10.1 months, though these rates were increased to 62% and 13.5 months respectively for patients with no prior HMA treatment103,104.
As with the azacitidine plus venetoclax success, a large, international phase 3 clinical trial, VIALE-C, was quick to follow105. Enrolled participants were ineligible for intensive chemotherapy and were treated either with venetoclax or placebo with LDAC. As of summer 2020, OS was 4.1 and 7.2 months and CR rates were 13% and 48% in the control and venetoclax arms, respectively106. This trial continues in follow-up, but in 2020 these promising initial results led to FDA full approval of LDAC with venetoclax in treatment-naive AML patients.
Further pre-clinical combinations of conventional therapy with BH3 mimetics in AML
The success of venetoclax in AML in combination with the standard-of-care therapies HMA and LDAC has led to investigations into combining this BH3 mimetic with other clinically available therapies. Here we will briefly describe some of these pre-clinical investigations.
Midostaurin
The apoptotic response to the FLT3 kinase inhibitor midostaurin in FLT3-ITD-positive primary AML samples and cell lines is enhanced in the presence of venetoclax107. FLT3-ITD upregulates MCL-1 through STAT5 activation and the Akt pathway; therefore, inhibition of FLT3-ITD and treatment with venetoclax concomitantly removes the protection of both MCL-1 and BCL-2, rendering the cell sensitive to apoptosis58,108.
Sorafenib
Sorafenib is a multi-kinase inhibitor targeting RAF, PDGFRB, VEGFR2, FLT3, and KIT, and induces apoptosis in AML cells via BIM and downregulation of MCL-1109,110. Further sensitizing cells to BIM with BH3 mimetics potentiates the apoptotic effect of sorafenib, as seen in combination with obatoclax and venetoclax111.
All-trans retinoic acid (ATRA)
MCL-1 overexpression impedes the ability of ATRA to induce growth arrest and differentiation in APL and combining ATRA with an MCL-1-interfering BH3 mimetic has been postulated to induce a greater cytotoxic response than ATRA alone112. However, the combination of JY-1-106 with ATRA was shown in one study to have little effect on reducing cell proliferation in HL-60, an APL cell line113.
Daunorubicin
Daunorubicin is a DNA-intercalating chemotherapeutic able to induce sphingomyelin hydrolysis and ceramide generation114. Overexpression of BCL-2 has been shown to prevent daunorubicin-induced apoptosis in AML cell lines through inhibition of X-linked inhibitor of apoptosis protein (XIAP) and degradation of Akt115,116. Removal of this BCL-2-mediated protection against daunorubicin has been shown to be effective at synergistically inducing apoptosis and growth inhibition in cell lines and in patient samples, using either ABT-737 or venetoclax71,117.
Combining mimetics
The possibility of combining BH3 mimetics with different target specificities is also under scrutiny in both CML and AML studies, although toxicity concerns have potentially held back investigations of this nature70,118,119. Combining BH3 mimetics has the advantage of disabling the cell’s ability to ‘switch’ between prosurvival proteins, a commonly reported resistance mechanism to BCL-2 inhibition, and thus overcoming the redundancy in the BCL-2 family system70,120,121,122.
Further clinical trials with BH3 mimetics in AML
In contrast to the clinical success of venetoclax, clinical trials of MCL-1 inhibitors have been more problematic. Initially, there were difficulties in developing MCL-1 inhibitors as the binding site is shallower and less flexible than that of BCL-2 or BCL-xL123. Recently, however, 4 agents (S64315124, AMG176125, AMG397126, and AZD5991127) with activity against MCL-1 entered phase 1 clinical trials as single agents in AML (NCT02979366128, NCT02675452129, NCT03465540130, NCT03218683131, with a view to combining with venetoclax (NCT03672695 132, NCT03797261133, NCT03218683131) or azacitidine (NCT02675452129), once dose-finding studies are completed. Importantly, CDK9 inhibitors (e.g., alvocidib, dinaciclib, CYC065, and AZD4573134,135,136,137) indirectly inhibit MCL-1. These agents have preclinical activity in AML, and a number of early phase clinical trials are ongoing. It will be important to determine if they have efficacy with a favorable safety profile.
The BCL-2/BCL-xL inhibitor navitoclax has undergone extensive clinical trial evaluation in solid tumor, lymphoid malignancies and myeloproliferative neoplasms, but not AML. Further development has been limited by the predicted and on-target side effect of thrombocytopenia67.
Measuring BCL-2 family dependence
Heterogenous responses to BH3 mimetics occurs in patients, indicating a need for personalizing treatment approaches when considering these drugs. BH3 profiling, a technique developed to predict relative dependency on BCL-2, MCL-1, and BCL-xL, has been shown to be useful in predicting responses of patients with AML after treatment with venetoclax, as well as highlighting potential resistance mechanisms88.
The success of BH3 profiling in this regard has led to the incorporation of this technique in several clinical trials as a prognostic marker and determinant of response, most notably for myelodysplastic syndrome and AML138,139,140,141, but also in trials focusing on CLL142 and ALL143. Further, combining BH3 profiling results with basal expression data for the prosurvival BCL-2 proteins (termed ‘mitochondrial profiling’) has also been shown to be effective in indicating BCL-2 dependence144. In the case of the clinical trial NCT02520011145, demonstrable MCL-1 dependence in AML, as determined by mitochondrial profiling, was used to identify eligible patients, although this trial was later terminated due to slow enrollment.
In addition, protein and gene expression profiles of the target BCL-2 family members146,147 and expression of BH3-only proteins such as BIM148 have been shown to correlate with response to BH3 mimetics.
BH3 and mitochondrial profiling, along with gene and protein expression data, represent high-throughput methods with a fast turnaround time, often requiring few cells and with limited ex vivo exposure of patient samples. As these techniques are refined, there is a trend in the literature towards assays with high specificity and sensitivity for identifying patients who may benefit from BH3 mimetic treatment, warranting investigations into the point-of-care applications of these assays.
Future directions
With substantial progress being made in the field of BCL-2-targeted therapies and our increasing understanding of dysregulation of this family in the myeloid leukemias, great strides have been made in bringing these areas together, as highlighted in this review.
A number of unanswered questions and areas for further investigation remain to be addressed. With the difficulties in targeting MCL-1 and BCL-xL, the identification of a therapeutic window is required, which could be addressed through sequential or alternating treatment strategies to allow time for healthy tissue recovery, or through potent combinations that would allow for a substantial reduction in the concentrations of BH3 mimetic required. Further, measuring BCL-2 family dependency in a point-of-care setting to refine treatment deserves additional scrutiny to determine clinical utility.
As clinical trials advance and standard treatment regimens incorporate BH3 mimetics to a greater degree, these novel therapeutic combinations may represent a significant step in the direction of targeted, personalized therapy for patients with myeloid leukemias.
References
Kerr, J. F. R., Wyllie, A. H. & Currie, A. R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Kroemer, G. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11 (2009).
Ashkenazi, A. Targeting the extrinsic apoptotic pathway in cancer: lessons learned and future directions. J. Clin. Invest. 125, 487–489 (2015).
Vaux, D. L., Weissman, I. L. & Kim, S. K. Prevention of programmed cell death in Caenorhabditis elegans by human BCL-2. Science 258, 1955–1957 (1992).
Oberst, A., Bender, C. & Green, D. R. Living with death: the evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 15, 1139–1146 (2008).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Singh, R., Letai, A. & Sarosiek, K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 20, 175–193 (2019).
Tsujimoto, Y., Cossman, J., Jaffe, E. & Croce, C. M. Involvement of the BCL-2 gene in human follicular lymphoma. Science 228, 1440–1443 (1985).
Pan, R. et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 4, 362–375 (2014).
Campos, L. et al. High expression of BCL-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 81, 3091–3096 (1993).
Aichberger, K. J. et al. Low-level expression of proapoptotic BCL-2-interacting mediator in leukemic cells in patients with chronic myeloid leukemia: role of BCR/ABL, characterization of underlying signaling pathways, and reexpression by novel pharmacologic compounds. Cancer Res. 65, 9436–9444 (2005).
Aichberger, K. J. et al. Identification of MCL-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and MCL-1 antisense oligonucleotides. Blood 105, 3303–3311 (2005).
Jaiswal, S. et al. Expression of BCR/ABL and BCL-2 in myeloid progenitors leads to myeloid leukemias. Proc. Natl Acad. Sci. USA 100, 10002–10007 (2003).
Handa, H. et al. Bcl-2 and c-myc expression, cell cycle kinetics and apoptosis during the progression of chronic myelogenous leukemia from diagnosis to blastic phase. Leuk. Res. 21, 479–489 (1997).
Sánchez-García, I. & Grütz, G. Tumorigenic activity of the BCR-ABL oncogenes is mediated by BCL2. Proc. Natl Acad. Sci. USA 92, 5287–5291 (1995).
Kline, M. P. et al. ABT-737, an inhibitor of BCL-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia 21, 1549–1560 (2007).
Shivakumar, L. & Armitage, J. O. BCL-2 gene expression as a predictor of outcome in diffuse large B-cell lymphoma. Clin. Lymphoma Myeloma 6, 455–457 (2006).
Coustan-Smith, E. et al. Clinical relevance of BCL-2 overexpression in childhood acute lymphoblastic leukemia. Blood 87, 1140–1146 (1996).
Youle, R. J. & Strasser, A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 (2008).
Westphal, D., Dewson, G., Czabotar, P. E. & Kluck, R. M. Molecular biology of Bax and Bak activation and action. Biochim Biophys. Acta - Mol. Cell Res. 1813, 521–531 (2011).
Riedl, S. J. & Salvesen, G. S. The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 8, 405–413 (2007).
Letai, A. et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2, 183–192 (2002).
Bock, F. J. & Tait, S. W. G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100 (2020).
Chen, L. et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17, 393–403 (2005).
Delbridge, A. R. D. & Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 22, 1071–1080 (2015).
Shtivelman, E., Lifshitz, B., Gale, R. P. & Canaani, E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 315, 550–554 (1985).
Ben-Neriah, Y., Daley, G. Q., Mes-Masson, A.-M., Witte, O. N. & Baltimore, D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr-abl hybrid gene. Science 233, 212–214 (1986).
Holyoake, T., Jiang, X., Eaves, C. & Eaves, A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 94, 2056–2064 (1999).
Deininger, M. W. N., Goldman, J. M., Lydon, N. & Melo, J. V. The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood 90, 3691–3698 (1997).
Hehlmann, R. How I treat CML blast crisis. Blood 120, 737–747 (2012).
Kuribara, R. et al. Roles of Bim in apoptosis of normal and Bcr-Abl-expressing hematopoietic progenitors. Mol. Cell Biol. 24, 6172–6183 (2004).
Kuroda, J. et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc. Natl Acad. Sci. USA 103, 14907–14912 (2006).
Amarante-Mendes, G. et al. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome C and activation of caspase-3. Blood 91, 1700–1705 (1998).
Gesbert, F. & Griffin, J. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood 96, 2269 (2000).
Carter, B. Z. et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci. Transl. Med. 8, 355ra117 (2016).
Kawauchi, K., Ogasawara, T., Yasuyama, M. & Ohkawa, S. I. Involvement of Akt kinase in the action of STI571 on chronic myelogenous leukemia cells. Blood Cells, Mol. Dis. 31, 11–17 (2003).
Yamaguchi, H. & Wang, H. G. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 20, 7779–7786 (2001).
Keeshan, K., Cotter, T. & McKenna, S. High Bcr-Abl expression prevents the translocation of Bax and Bad to the mitochondrion. Leukemia 16, 1725–1734 (2002).
Li, Y. et al. MiR-29b suppresses CML cell proliferation and induces apoptosis via regulation of BCR/ABL1 protein. Exp. Cell Res. 319, 1094–1101 (2013).
Machová Polaková, K. et al. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol. Cancer 10, 1–13 (2011).
Short, N. J., Rytting, M. E. & Cortes, J. E. Acute myeloid leukaemia. Lancet 392, 593–606 (2018).
Döhner, H. et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447 (2017).
Saultz, J. & Garzon, R. Acute myeloid leukemia: a concise review. J. Clin. Med. 5, 33 (2016).
Di Nardo, C. D. & Cortes, J. E. Mutations in AML: prognostic and therapeutic implications. Hematology 2016, 348–355 (2016).
Stone, R. M. et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 377, 454–464 (2017).
Andreeff, M. et al. Expression of Bcl-2-related genes in normal and AML progenitors: changes induced by chemotherapy and retinoic acid. Leukemia 13, 1881–1892 (1999).
Letai, A., Sorcinelli, M. D., Beard, C. & Korsmeyer, S. J. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 6, 241–249 (2004).
Zhou, J. D. et al. BCL2 overexpression: clinical implication and biological insights in acute myeloid leukemia. Diagn. Pathol. 14, 68 (2019).
Bensi, L. et al. Bcl-2 oncoprotein expression in acute myeloid leukemia. Haematologica 80, 98–102 (1995).
Lauria, F. et al. High BCL-2 expression in acute myeloid leukemia cells correlates with CD34 positivity and complete remission rate. Leukemia 11, 2075–2078 (1997).
Wang, J. et al. Cell permeable Bcl-2 binding peptides: a chemical approach to apoptosis induction in tumor cells. Cancer Res. 60, 1498–1502 (2000).
Wang, J. L. et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl Acad. Sci. USA 97, 7124–7129 (2000).
Pallis, M., Zhu, Y. M. & Russell, N. H. Bcl-x(L) is heterogeneously expressed by acute myeloblastic leukaemia cells and is associated with autonomous growth in vitro and with P-glycoprotein expression. Leukemia 11, 945–949 (1997).
Konopleva, M. et al. The anti-apoptotic genes Bcl-X(L) and Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+ cells. Br. J. Haematol. 118, 521–534 (2002).
Kaufmann, S. H. et al. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 91, 991–1000 (1998).
Xiang, Z. et al. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J. Clin. Invest. 120, 2109–2118 (2010).
Glaser, S. P. et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 26, 120–125 (2012).
Yoshimoto, G. et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 114, 5034–5043 (2009).
Wei, A. H. et al. Targeting MCL-1 in hematologic malignancies: rationale and progress. Blood Rev. 44, 100672 (2020).
Ryan, J., Montero, J., Rocco, J. & Letai, A. iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol. Chem. 397, 671–678 (2016).
Ryan, J. & Letai, A. BH3 profiling in whole cells by fluorimeter or FACS. Methods 61, 156–164 (2013).
Lessene, G., Czabotar, P. E. & Colman, P. M. BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 7, 989–1000 (2008).
Leverson, J. D. et al. Found in translation: how preclinical research is guiding the clinical development of the BCL2-selective inhibitor venetoclax. Cancer Discov. 7, 1376–1393 (2017).
Oltersdorf, T. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 (2005).
Tse, C. et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, 3421–3428 (2008).
Zhang, H. et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 14, 943–951 (2007).
Roberts, A. W. et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 30, 488–496 (2012).
Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 (2013).
Choudhary, G. S. et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 6, e1593–e1593 (2015).
Lin, K. H. et al. Targeting MCL-1/BCL-XL forestalls the acquisition of resistance to ABT-199 in acute myeloid leukemia. Sci. Rep. 6, 1–10 (2016).
Niu, X. et al. Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin. Cancer Res. 22, 4440–4451 (2016).
Vo, T. T. et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 151, 344–355 (2012).
Schoenwaelder, S. M. et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 118, 1663–1674 (2011).
Wang, X. et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 27, 1351–1364 (2013).
Thomas, R. L. et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 27, 1365–1377 (2013).
Graham, S. M. et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 99, 319–325 (2002).
Holyoake, T. L. & Vetrie, D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood 129, 1595–1606 (2017).
Ng, K. P. et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 18, 521–528 (2012).
La Rosée, P. & Deininger, M. W. Resistance to imatinib: mutations and beyond. Semin Hematol. 47, 335–343 (2010).
Ko, T. K., CTHH, Chuah & JWJJ, Huang Ng K-PP, Ong ST. The BCL2 inhibitor ABT-199 significantly enhances imatinib-induced cell death in chronic myeloid leukemia progenitors. Oncotarget 5, 9033–9038 (2014).
Mak, D. H. et al. Activation of apoptosis signaling eliminates CD34+ progenitor cells in blast crisis CML independent of response to tyrosine kinase inhibitors. Leukemia 26, 788–794 (2012).
Airiau, K. et al. ABT-737 increases tyrosine kinase inhibitor-induced apoptosis in chronic myeloid leukemia cells through XIAP downregulation and sensitizes CD34+ CD38- population to imatinib. Exp. Hematol. 40, 367–78.e2 (2012).
Dasatinib and venetoclax in treating patients with Philadelphia chromosome positive or BCR-ABL1 positive early chronic phase chronic myelogenous leukemia. https://clinicaltrials.gov/ct2/show/NCT02689440 (2020).
Decitabine, venetoclax, and ponatinib for the treatment of Philadelphia chromosome-positive acute myeloid leukemia or myeloid blast phase chronic myelogenous leukemia https://clinicaltrials.gov/ct2/show/NCT04188405 (2020).
Niu, X. et al. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia 28, 1557–1560 (2014).
Chan, S. M. et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 21, 178–184 (2015).
A phase 2 study of ABT-199 in subjects with acute myelogenous leukemia (AML) https://clinicaltrials.gov/ct2/show/NCT01994837 (2021).
Konopleva, M. et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 6, 1106–1117 (2016).
Pollyea, D. A., Amaya, M., Strati, P. & Konopleva, M. Y. Venetoclax for AML: changing the treatment paradigm. Blood Adv. 3, 4326–4335 (2019).
Stoetzer, O. J. et al. Association of BCL-2, BAX, BCL-xL and interleukin-1 beta-converting enzyme expression with initial response to chemotherapy in acute myeloid leukemia. Leukemia 10, S18–22 (1996). Suppl 3.
Karakas, T. et al. High expression of BCL-2 mRNA as a determinant of poor prognosis in acute myeloid leukemia. Ann. Oncol. 9, 159–165 (1998).
Bogenberger, J. M. et al. BCL-2 family proteins as 5-azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 28, 1657–1665 (2014).
Bogenberger, J. M. et al. Ex vivo activity of BCL-2 family inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk. Lymphoma 56, 226–229 (2015).
Tsao, T. et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann. Hematol. 91, 1861–1870 (2012).
Study of ABT-199 (GDC-0199) in combination with azacitidine or decitabine (chemo combo) in subjects with acute myelogenous leukemia (AML). https://clinicaltrials.gov/ct2/show/NCT02203773 (2021).
DiNardo, C. D. et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 19, 216–228 (2018).
DiNardo, C. D. et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 133, 7–17 (2019).
A study of venetoclax in combination with azacitidine versus azacitidine in treatment naïve subjects with acute myeloid leukemia who are ineligible for standard induction therapy https://clinicaltrials.gov/ct2/show/NCT02993523 (2021).
DiNardo, C. D. et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 383, 617–629 (2020).
Benedict, W. F., Harris, N. & Karon, M. Kinetics of 1-β-d-arabinofuranosylcytosine-induced chromosome breaks. Cancer Res. 30, 2477–2483 (1970).
Keith, F. J., Bradbury, D. A., Zhu, Y. M. & Russell, N. H. Inhibition of BCL-2 with antisense oligonucleotides induces apoptosis and increases the sensitivity of AML blasts to Ara-C. Leukemia 9, 131–138 (1995).
Xie, C. et al. Obatoclax potentiates the cytotoxic effect of cytarabine on acute myeloid leukemia cells by enhancing DNA damage. Mol. Oncol. 9, 409–421 (2015).
A study evaluating venetoclax in combination with low-dose cytarabine in treatment-naïve participants with acute myelogenous leukemia https://clinicaltrials.gov/ct2/show/NCT02287233 (2021).
Wei, A. H. et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J. Clin. Oncol. 37, 1277–1284 (2019).
A study of venetoclax in combination with low dose cytarabine versus low dose cytarabine alone in treatment naive patients with acute myeloid leukemia who are ineligible for intensive chemotherapy. https://clinicaltrials.gov/ct2/show/NCT03069352 (2021).
Wei, A. H. et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood 135, 2137–2145 (2020).
Ma, J. et al. Inhibition of Bcl-2 synergistically enhances the antileukemic activity of midostaurin and gilteritinib in preclinical models of FLT3-mutated acute myeloid leukemia. Clin. Cancer Res. 25, 6815–6826 (2019).
Seipel, K., Marques, M. A. T., Sidler, C., Mueller, B. U. & Pabst, T. MDM2- and FLT3-inhibitors in the treatment of FLT3-ITD acute myeloid leukemia, specificity and efficacy of NVP-HDM201 and midostaurin. Haematologica 103, 1862–1872 (2018).
Zhang, W. et al. Sorafenib induces apoptosis of AML cells via Bim-mediated activation of the intrinsic apoptotic pathway. Leukemia 22, 808–818 (2008).
Rahmani, M., Davis, E. M., Bauer, C., Dent, P. & Grant, S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J. Biol. Chem. 280, 35217–35227 (2005).
Rahmani, M. et al. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood 119, 6089–6098 (2012).
Yang, J., Ikezoe, T., Nishioka, C. & Yokoyama, A. Over-expression of Mcl-1 impairs the ability of ATRA to induce growth arrest and differentiation in acute promyelocytic leukemia cells. Apoptosis 18, 1403–1415 (2013).
Perri, M. et al. BCL-xL/MCL-1 inhibition and RARγ antagonism work cooperatively in human HL60 leukemia cells. Exp. Cell Res. 327, 183–191 (2014).
Jaffrézou, J. P. et al. Daunorubicin-induced apoptosis: triggering of ceramide generation through sphingomyelin hydrolysis. EMBO J. 15, 2417–2424 (1996).
Kim, Y. H., Park, J. W., Lee, J. Y., Surh, Y. J. & Kwon, T. K. Bcl-2 overexpression prevents daunorubicin-induced apoptosis through inhibition of XIAP and Akt degradation. Biochem. Pharmacol. 66, 1779–1786 (2003).
Allouche, M. et al. Influence of Bcl-2 overexpression on the ceramide pathway in daunorubicin-induced apoptosis of leukemic cells. Oncogene 14, 1837–1845 (1997).
Dariushnejad, H. et al. ABT-737, synergistically enhances daunorubicin-mediated apoptosis in acute myeloid leukemia cell lines. Adv. Pharm. Bull. 4, 185–189 (2014).
Moujalled, D. M. et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 33, 905–917 (2019).
Carter, B. Z. et al. TP53 deficient/mutant AMLs are resistant to individual BH3 mimetics: high efficacy of combined inhibition of Bcl-2 and Mcl-1. Blood 134, 1271 (2019).
Yecies, D., Carlson, N. E., Deng, J. & Letai, A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 115, 3304–3313 (2010).
Fresquet, V., Rieger, M., Carolis, C., García-Barchino, M. J. & Martinez-Climent, J. A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 123, 4111–4119 (2014).
Hormi, M. et al. Pairing MCL‐1 inhibition with venetoclax improves therapeutic efficiency of BH3‐mimetics in AML. Eur. J. Haematol. 105, 588–596 (2020).
Wan, Y., Dai, N., Tang, Z. & Fang, H. Small-molecule Mcl-1 inhibitors: emerging anti-tumor agents. Eur. J. Med. Chem. 146, 471–482 (2018).
Szlávik, Z. et al. Structure-guided discovery of a selective MCL-1 inhibitor with cellular activity. J. Med. Chem. 62, 6913–6924 (2019).
Yi, X. et al. AMG-176, an Mcl-1 antagonist, shows preclinical efficacy in chronic lymphocytic leukemia. Clin. Cancer Res. 26, 3856–3867 (2020).
Hird, A. W. & Tron, A. E. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharm. Ther. 198, 59–67 (2019).
Tron, A. E. et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 9, 5341 (2018).
Phase I study of S64315 administred intravenously in patients with acute myeloid leukaemia or myelodysplastic syndrome. https://clinicaltrials.gov/ct2/show/NCT02979366 (2020).
AMG 176 first in human trial in subjects with relapsed or refractory multiple myeloma and subjects with relapsed or refractory acute myeloid leukemia. https://clinicaltrials.gov/ct2/show/NCT02675452 (2020).
Safety, tolerability, pharmacokinetics and efficacy of AMG 397 in subjects with multiple myeloma, NHL, and AML. https://clinicaltrials.gov/ct2/show/NCT03465540 (2020).
Study of AZD5991 in relapsed or refractory haematologic malignancies. https://clinicaltrials.gov/ct2/show/NCT03218683 (2021).
Phase I dose escalation study of intravenously administered S64315 in combination with orally administered venetoclax in patients with acute myeloid leukaemia. https://clinicaltrials.gov/ct2/show/NCT03672695 (2020).
A study of venetoclax and AMG 176 in patients with relapsed/refractory hematologic malignancies. https://clinicaltrials.gov/ct2/show/NCT03797261 (2020).
Dasmahapatra, G., Almenara, J. A. & Grant, S. Flavopiridol and histone deacetylase inhibitors promote mitochondrial injury and cell death in human leukemia cells that overexpress Bcl-2. Mol. Pharmacol. 69, 288–298 (2006).
Baker, A. et al. The CDK9 inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of MLL-rearranged acute myeloid leukemia. Cancer Res. 76, 1158–1169 (2016).
Chantkran, W., Zheleva, D., Frame, S., Hsieh, Y.-C. & Copland, M. Combination of CYC065, a second generation CDK2/9 inhibitor, with venetoclax or standard chemotherapies - a novel therapeutic approach for acute myeloid leukaemia (AML). Blood 134, 3938–3938 (2019).
Cidado, J. et al. AZD4573 is a highly selective CDK9 inhibitor that suppresses Mcl-1 and induces apoptosis in hematologic cancer cells. Clin. Cancer Res. 26, 922–934 (2020).
Ivosidenib and venetoclax with or without azacitidine in treating participants with IDH1 mutated hematologic malignancies. https://clinicaltrials.gov/ct2/show/NCT03471260?term=bh3+profiling&draw=2&rank=8 (2020).
Venetoclax with combination chemotherapy in treating patients with newly diagnosed or relapsed or refractory acute myeloid leukemia. https://clinicaltrials.gov/ct2/show/NCT03214562?term=bh3+profiling&draw=2&rank=6 (2020).
A phase 1b/2 study of alvocidib plus decitabine or azacitidine in patients with MDS. https://clinicaltrials.gov/ct2/show/NCT03593915?term=bh3+profiling&draw=2&rank=2 (2020).
Venetoclax and decitabine in treating participants with relapsed/refractory acute myeloid leukemia or relapsed high-risk myelodysplastic syndrome. https://clinicaltrials.gov/ct2/show/NCT03404193?term=bh3+profiling&draw=2&rank=5 (2020).
Ibrutinib and venetoclax in treating patients with chronic lymphocytic leukemia after ibrutinib resistance. https://clinicaltrials.gov/ct2/show/NCT03943342?term=bh3+profiling&draw=2&rank=7 (2020).
Everolimus with multiagent re-induction chemotherapy in pediatric patients with ALL. https://clinicaltrials.gov/ct2/show/NCT01523977?term=bh3+profiling&draw=2&rank=4 (2020).
Ishizawa, J. et al. Mitochondrial profiling of acute myeloid leukemia in the assessment of response to apoptosis modulating drugs. PLoS ONE 10, e0138377 (2015).
Alvocidib Biomarker-driven Phase 2 AML Study. https://clinicaltrials.gov/ct2/show/NCT02520011?term=NCT02520011&draw=2&rank=1 (2020).
Punnoose, E. A. et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol. Cancer Ther. 15, 1132–1144 (2016).
Soderquist, R. S. et al. Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nat. Commun. 9, 3513 (2018).
Iavarone, C. et al. Combined MEK and Bcl-2/XL inhibition is effective in high-grade serous ovarian cancer patient–derived xenograft models and BIM levels are predictive of responsiveness. Mol. Cancer Ther. 18, 642–655 (2019).
Acknowledgements
The authors would like to thank the University of Glasgow and the Wellcome Trust Institutional Strategic Support Fund [204820/Z/16/Z] for supporting this work.
Funding statement
This work was funded by the University of Glasgow and the Wellcome Trust Institutional Strategic Support Fund [204820/Z/16/Z].
Author information
Authors and Affiliations
Contributions
N.P. performed writing and revisions of the paper. M.C. and H.W. provided writing and review. All authors contributed to the concept. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This work required no ethical approval.
Conflict of interest
M.C. has received research funding from Novartis, Bristol-Myers Squibb, Cyclacel and Takeda/Incyte, is/has been an advisory board member for Bristol-Myers Squibb, Novartis, Incyte, Daiichi Sankyo, and Pfizer and has received honoraria from Astellas, Bristol-Myers Squibb, Novartis, Incyte, Pfizer and Gilead. N.P. and H.W. have no conflicts-of-interest to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by F. Pentimalli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Parry, N., Wheadon, H. & Copland, M. The application of BH3 mimetics in myeloid leukemias. Cell Death Dis 12, 222 (2021). https://doi.org/10.1038/s41419-021-03500-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03500-6
This article is cited by
-
Regulation of anoikis by extrinsic death receptor pathways
Cell Communication and Signaling (2023)
-
Discovery and identification of a novel small molecule BCL-2 inhibitor that binds to the BH4 domain
Acta Pharmacologica Sinica (2023)
-
The miR-106b-25 cluster mediates drug resistance in myeloid leukaemias by inactivating multiple apoptotic genes
International Journal of Hematology (2023)
-
BH3 mimetics in combination with nilotinib or ponatinib represent a promising therapeutic strategy in blast phase chronic myeloid leukemia
Cell Death Discovery (2022)
-
Artesunate improves venetoclax plus cytarabine AML cell targeting by regulating the Noxa/Bim/Mcl-1/p-Chk1 axis
Cell Death & Disease (2022)
