
Abstract
Cytoplasmic Janus protein tyrosine kinases (JAKs) are crucial components of diverse signal transduction pathways that govern cellular survival, proliferation, differentiation and apoptosis. Evidence to date, indicates that JAK kinase function may integrate components of diverse signaling cascades. While it is likely that activation of STAT proteins may be an important function attributed to the JAK kinases, it is certainly not the only function performed by this key family of cytoplasmic tyrosine kinases. Emerging evidence indicates that phosphorylation of cytokine and growth factor receptors may be the primary functional attribute of JAK kinases. The JAK-triggered receptor phosphorylation can potentially be a rate-limiting event for a successful culmination of downstream signaling events. In support of this hypothesis, it has been found that JAK kinase function is required for optimal activation of the Src-kinase cascade, the Ras-MAP kinase pathway, the PI3K-AKT pathway and STAT signaling following the interaction of cytokine/interferon receptors with their ligands. Aberrations in JAK kinase activity, that may lead to derailment of one or more of the above mentioned pathways could disrupt normal cellular responses and result in disease states. Thus, over-activation of JAK kinases has been implicated in tumorigenesis. In contrast, loss of JAK kinase function has been found to result in disease states such as severe-combined immunodeficiency. In summary, optimal JAK kinase activity is a critical determinant of normal transmission of cytokine and growth factor signals.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

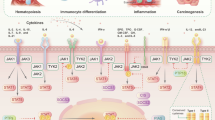

References
Adams JM and Cory S. . 1998 Science 281: 1322–1326.
Al-Shami A and Naccache PH. . 1999 J. Biol. Chem. 274: 5333–5338.
Alam R, Pazdrak K, Stafford S and Forsythe P. . 1995 Int. Arch. Allergy Immunol. 107: 226–227.
Antonsson B and Martinou JC. . 2000 Exp. Cell. Res. 256: 50–57.
Argetsinger LS, Campbell GS, Yang X, Witthuhn BA, Silvennoinen O, Ihle JN and Carter-Su C. . 1993 Cell 74: 237–244.
Azam M, Erdjument-Bromage H, Kreider BL, Xia M, Quelle F, Basu R, Saris C, Tempst P, Ihle JN and Schindler C. . 1995 EMBO J. 14: 1402–1411.
Bacon CM, McVicar DW, Ortaldo JR, Rees RC, O'Shea JJ and Johnston JA. . 1995 J. Exp. Med. 181: 399–404.
Bates ME, Bertics PJ and Busse WW. . 1996 J. Immunol. 156: 711–718.
Binari R and Perrimon N. . 1994 Genes Dev. 8: 300–312.
Bittorf T, Seiler J, Zhang Z, Jaster R and Brock J. . 1999 Biol. Chem. 380: 1201–1209.
Briscoe J, Rogers NC, Witthuhn BA, Watling D, Harpur AG, Wilks AF, Stark GR, Ihle JN and Kerr IM. . 1996 EMBO J. 15: 799–809.
Cacalano NA, Migone TS, Bazan F, Hanson EP, Chen M, Candotti F, O'Shea JJ and Johnston JA. . 1999 EMBO J. 18: 1549–1558.
Caldenhoven E, van Dijk T, Raaijmakers JA, Lammers JW, Koenderman L and De Groot RP. . 1995 J. Biol. Chem. 270: 25778–25784.
Callus BA and Mathey-Prevot B. . 1998 Blood 91: 3182–3192.
Candotti F, Oakes SA, Johnston JA, Giliani S, Schumacher RF, Mella P, Fiorini M, Ugazio AG, Badolato R, Notarangelo LD, Bozzi F, Macchi P, Strina D, Vezzoni P, Blaese RM, O'Shea JJ and Villa A. . 1997 Blood 90: 3996–4003.
Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, Drago J, Noguchi M, Grinberg A, Bloom ET, Paul WE, Katz SI, Love PE and Leonard WJ. . 1995 Immunity 2: 223–238.
Cao X, Tay A, Guy GR and Tan YH. . 1996 Mol. Cell. Biol. 16: 1595–1603.
Carlesso N, Frank DA and Griffin JD. . 1996 J. Exp. Med. 183: 811–820.
Chaturvedi P, Reddy MV and Reddy EP. . 1998 Oncogene 16: 1749–1758.
Chaturvedi P, Sharma S and Reddy EP. . 1997 Mol. Cell. Biol. 17: 3295–3304.
Chen M, Cheng A, Chen YQ, Hymel A, Hanson EP, Kimmel L, Minami Y, Taniguchi T, Changelian PS and O'Shea JJ. . 1997 Proc. Natl. Acad. Sci. USA 94: 6910–6915.
Colamonici O, Yan H, Domanski P, Handa R, Smalley D, Mullersman J, Witte M, Krishnan K and Krolewski J. . 1994a Mol. Cell. Biol. 14: 8133–8142.
Colamonici OR, Uyttendaele H, Domanski P, Yan H and Krolewski JJ. . 1994b J. Biol. Chem. 269: 3518–3522.
Cook WD, Metcalf D, Nicola NA, Burgess AW and Walker F. . 1985 Cell 41: 677–683.
Cooper JA and Howell B. . 1993 Cell 73: 1051–1054.
Copeland NG, Gilbert DJ, Schindler C, Zhong Z, Wen Z, Darnell Jr JE, Mui AL, Miyajima A, Quelle FW, Ihle JN and Jenkins NA. . 1995 Genomics 29: 225–228.
Cornelis S, Fache I, Van der Heyden J, Guisez Y, Tavernier J, Devos R, Fiers W and Plaetinck G. . 1995 Eur. J. Immunol. 25: 1857–1864.
Danial NN, Pernis A and Rothman PB. . 1995 Science 269: 1875–1877.
Danial NN, Losman JA, Lu T, Yip N, Krishnan K, Krolewski J, Goff SP, Wang JY and Rothman PB. . 1998 Mol. Cell. Biol. 18: 6795–6804.
Darnell Jr JE. . 1997 Science 277: 1630–1635.
Darnell Jr JE. . 1998 J. Interferon Cytokine Res. 18: 549–554.
Darnell Jr JE, Kerr IM and Stark GR. . 1994 Science 264: 1415–1421.
DaSilva L, Howard OM, Rui H, Kirken RA and Farrar WL. . 1994 J. Biol. Chem. 269: 18267–18270.
David M, Chen HE, Goelz S, Larner AC and Neel BG. . 1995 Mol. Cell. Biol. 15: 7050–7058.
DiSanto JP, Muller W, Guy-Grand D, Fischer A and Rajewsky K. . 1995 Proc. Natl. Acad. Sci. USA 92: 377–381.
Duhe RJ and Farrar WL. . 1995 J. Biol. Chem. 270: 23084–23089.
Dusanter-Fourt I, Muller O, Ziemiecki A, Mayeux P, Drucker B, Djiane J, Wilks A, Harpur AG, Fischer S and Gisselbrecht S. . 1994 EMBO J. 13: 2583–2591.
Eder M, Ernst TJ, Ganser A, Jubinsky PT, Inhorn R, Hoelzer D and Griffin JD. . 1994 J. Biol. Chem. 269: 30173–30180.
Egan SE and Weinberg RA. . 1993 Nature 365: 781–783.
Eilers A, Kanda K, Klose B, Krolewski J and Decker T. . 1996 Cell Growth Differ. 7: 833–840.
Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S and Yoshimura A. . 1997 Nature 387: 921–924.
Feng J, Witthuhn BA, Matsuda T, Kohlhuber F, Kerr IM and Ihle JN. . 1997 Mol. Cell. Biol. 17: 2497–2501.
Firmbach-Kraft I, Byers M, Shows T, Dalla-Favera R and Krolewski JJ. . 1990 Oncogene 5: 1329–1336.
Fischer EH, Charbonneau H and Tonks NK. . 1991 Science 253: 401–406.
Flores-Morales A, Pircher TJ, Silvennoinen O, Gustafsson JA, Sanchez-Gomez M, Norstedt G, Haldosen LA and Wood TJ. . 1998 Mol. Cell. Endocrinol. 138: 1–10.
Frank SJ, Yi W, Zhao Y, Goldsmith JF, Gilliland G, Jiang J, Sakai I and Kraft AS. . 1995 J. Biol. Chem. 270: 14776–14785.
Frank DA and Varticovski L. . 1996 Leukemia 10: 1724–1780.
Gauzzi MC, Barbieri G, Richter MF, Uze G, Ling L, Fellous M and Pellegrini S. . 1997 Proc. Natl. Acad. Sci. USA 94: 11839–11844.
Gauzzi MC, Velazquez L, McKendry R, Mogensen KE, Fellous M and Pellegrini S. . 1996 J. Biol. Chem. 271: 20494–20500.
Giorgetti-Peraldi S, Peyrade F, Baron V and Van Obberghen E. . 1995 Eur. J. Biochem. 234: 656–660.
Goodman PA, Niehoff LB and Uckun FM. . 1998 J. Biol. Chem. 273: 17742–17748.
Goujon L, Allevato G, Simonin G, Paquereau L, Le Cam A, Clark J, Nielsen JH, Djiane J, Postel-Vinay MC, Edery M and Kelly PA. . 1994 Proc. Natl. Acad. Sci. USA 91: 957–961.
Grossman WJ, Verbsky JW, Yang L, Berg LJ, Fields LE, Chaplin DD and Ratner L. . 1999 Blood 94: 932–939.
Gurney AL, Wong SC, Henzel WJ and de Sauvage FJ. . 1995 Proc. Natl. Acad. Sci. USA 92: 5292–5296.
Guschin D, Rogers N, Briscoe J, Witthuhn B, Watling D, Horn F, Pellegrini S, Yasukawa K, Heinrich P, Stark GR, Ihle JN and Kerr IM. . 1995 EMBO J. 14: 1421–1429.
Hackett RH, Wang YD and Larner AC. . (1995). J. Biol. Chem. 270: 21326–21330.
Hanks SK, Quinn AM and Hunter T. . 1988 Science 241: 42–52.
Haque SJ, Harbor P, Tabrizi M, Yi T and Williams BR. . 1998 J. Biol. Chem. 273: 33893–33896.
Harpur AG, Andres AC, Ziemiecki A, Aston RR and Wilks AF. . 1992 Oncogene 7: 1347–1353.
Harrison DA, Binari R, Nahreini TS, Gilman M and Perrimon N. . 1995 EMBO J. 14: 2857–2865.
Heim MH. . 1999 J. Recept. Signal Transduct Res. 19: 75–120.
Horvath CM, Wen Z and Darnell Jr JE. . 1995 Genes Dev. 9: 984–994.
Igarashi K, Garotta G, Ozmen L, Ziemiecki A, Wilks AF, Harpur AG, Larner AC and Finbloom DS. . 1994 J. Biol. Chem. 269: 14333–14336.
Ihle JN, Nosaka T, Thierfelder W, Quelle FW and Shimoda K. . 1997 Stem Cells 15: 105–112.
Ihle JN, Witthuhn BA, Quelle FW, Yamamoto K and Silvennoinen O. . 1995 Ann. Rev. Immunol. 13: 369–398.
Ilaria Jr RL and Van Etten RA. . 1996 J. Biol. Chem. 271: 31704–31710.
Itoh T, Muto A, Watanabe S, Miyajima A, Yokota T and Arai K. . 1996 J. Biol. Chem. 271: 7587–7592.
Jiang N, He TC, Miyajima A and Wojchowski DM. . 1996 J. Biol. Chem. 271: 16472–16476.
Jiao H, Berrada K, Yang W, Tabrizi M, Platanias LC and Yi T. . 1996 Mol. Cell. Biol. 16: 6985–6992.
John J, McKendry R, Pellegrini S, Flavell D, Kerr IM and Stark GR. . 1991 Mol. Cell. Biol. 11: 4189–4195.
Johnston JA, Kawamura M, Kirken RA, Chen YQ, Blake TB, Shibuya K, Ortaldo JR, McVicar DW and O'Shea JJ. . 1994 Nature 370: 151–153.
Jubinsky PT, Nathan DG, Wilson DJ and Sieff CA. . 1993 Blood 81: 587–591.
Kawamura M, McVicar DW, Johnston JA, Blake TB, Chen YQ, Lal BK, Lloyd AR, Kelvin DJ, Staples JE, Ortaldo JR and O'Shea JJ. . 1994 Proc. Natl. Acad. Sci. USA 91: 6374–6378.
Kazansky AV, Kabotyanski EB, Wyszomierski SL, Mancini MA and Rosen JM. . 1999 J. Biol. Chem. 274: 22484–22492.
Kim TK and Maniatis T. . 1996 Science 273: 1717–1719.
Kirken RA, Rui H, Malabarba MG and Farrar WL. . 1994 J. Biol. Chem. 269: 19136–19141.
Kishimoto T, Taga T and Akira S. . 1994 Cell 76: 253–262.
Klingmuller U, Lorenz U, Cantley LC, Neel BG and Lodish HF. . 1995 Cell 80: 729–738.
Kohlhuber F, Rogers NC, Watling D, Feng J, Guschin D, Briscoe J, Witthuhn BA, Kono DH, Owens DG and Wechsler AR. . 1996 Mamm. Genome 7: 476–477.
Kotenko SV, Pestka S, Stark GR, Ihle JN and Kerr IM. . 1997 Mol. Cell. Biol. 17: 695–706.
Kotenko SV, Izotova LS, Pollack BP, Muthukumaran G, Paukku K, Silvennoinen O, Ihle JN and Pestka S. . 1996 J. Biol. Chem. 271: 17174–17182.
Kouro T, Kikuchi Y, Kanazawa H, Hirokawa K, Harada N, Shiiba M, Wakao H, Takaki S and Takatsu K. . 1996 Int. Immunol. 8: 237–245.
Krolewski JJ, Lee R, Eddy R, Shows TB and Dalla-Favera R. . 1990 Oncogene 5: 277–282.
Krishnan K, Pine R and Krolewski JJ. . 1997 Eur. J. Biochem. 274: 298–305.
Kruger A and Anderson SM. . 1991 Oncogene 6: 245–256.
Kumar A, Toscani A, Rane S and Reddy EP. . 1996 Oncogene 13: 2009–2014.
Kumar G, Gupta S, Wang S and Nel AE. . 1994 J. Immunol. 153: 4436–4447.
Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J and Bernard OA. . 1997 Science 278: 1309–1312.
Lai KS, Jin Y, Graham DK, Witthuhn BA, Ihle JN and Liu ET. . 1995 J. Biol. Chem. 270: 25028–25036.
Laneuville P, Heisterkamp N and Groffen J. . 1991 Oncogene 6: 275–282.
Leonard WJ. . 1996 Annu. Rev. Med. 47: 229–239.
Leonard WJ and Lin JX. . 2000 J. Allergy Clin. Immunol. 105: 877–888.
Leonard WJ, and O'Shea JJ. . 1998 Annu. Rev. Immunol. 16: 293–322.
Lin EY, Orlofsky A, Wang HG, Reed JC and Prystowsky MB. . 1996 Blood 87: 983–992.
Liu KD, Gaffen SL, Goldsmith MA and Greene WC. . 1997 Curr. Biol. 7: 817–826.
Luo H, Hanratty WP and Dearolf CR. . 1995 EMBO J. 14: 1412–1420.
Luo H, Rose P, Barber D, Hanratty WP, Lee S, Roberts TM, D'Andrea AD and Dearolf CR. . 1997 Mol. Cell. Biol. 17: 1562–1571.
Lutticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A, Harpur AG, Wilks AF, Yasukawa K, Taga T, Kishimoto T, Barbieri G, Pellegrini S, Sendtner M, Heinrich PC and Horn F. . 1994 Science 263: 89–92.
Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O'Shea JJ et al. 1995 Nature 377: 65–68.
Mathey-Prevot B, Nabel G, Palacios R and Baltimore D. . 1986 Mol. Cell. Biol. 6: 4133–4135.
Matsuguchi T, Inhorn RC, Carlesso N, Xu G, Druker B and Griffin JD. . 1995 EMBO J. 14: 257–265.
Matsuguchi T, Zhao Y, Lilly MB and Kraft AS. . 1997 J. Biol. Chem. 272: 17450–17459.
Matsumoto A, Masuhara M, Mitsui K, Yokouchi M, Ohtsubo M, Misawa H, Miyajima A and Yoshimura A. . 1997 Blood 89: 3148–3154.
McKendry R, John F, Flavell D, Muller M, Kerr IM and Stark GR. . 1991 Proc. Natl. Acad. Sci. USA 88: 11455–11459.
Migone TS, Cacalano NA, Taylor N, Yi T, Waldmann TA and Johnston JA. . 1998 Proc. Natl. Acad. Sci. USA 95: 3845–3850.
Miura O, Nakamura N, Quelle FW, Witthuhn BA, Ihle JM and Aoki N. . 1994 Blood 84: 1501–1507.
Mizuguchi R and Hatakeyama M. . 1998 J. Biol. Chem. 273: 32297–32303.
Mizuguchi R, Noto S, Yamada M, Ashizawa S, Higashi H and Hatakeyama M. . 2000 Jpn. J. Cancer Res. 91: 527–533.
Mui AL, Wakao H, Harada N, O'Farrell AM and Miyajima A. . 1995a J. Leukoc. Biol. 57: 799–803.
Mui AL, Wakao H, O'Farrell AM, Harada N and Miyajima A. . 1995b EMBO J. 14: 1166–1675.
Muller M, Briscoe J, Laxton C, Guschin D, Ziemiecki A, Silvennoinen O, Harpur AG, Barbieri G, Witthuhn BA, Schindler C, Pellegrini S, Wilks AF, Ihle JN, Stark GR and Kerr IM. . 1993 Nature 366: 129–135.
Naka T, Fujimoto M and Kishimoto T. . 1999 Trends Biochem. Sci. 24: 394–398.
Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S and Kishimoto T. . 1997 Nature 387: 924–929.
Nakamura N, Chin H, Miyasaka N and Miura O. . 1996 J. Biol. Chem. 271: 19483–19488.
Narazaki M, Fujimoto M, Matsumoto T, Morita Y, Saito H, Kajita T, Yoshizaki K, Naka T and Kishimoto T. . 1998 Proc. Natl. Acad. Sci. USA 95: 13130–13134.
Narazaki M, Witthuhn BA, Yoshida K, Silvennoinen O, Yasukawa K, Ihle JN, Kishimoto T and Taga T. . 1994 Proc. Natl. Acad. Sci. USA 91: 2285–2289.
Nelson BH, Lord JD and Greenberg PD. . 1996 Mol. Cell. Biol. 16: 309–317.
Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U and Pfeffer K. . 1998 Cell 93: 397–409.
Nicholson SE and Hilton DJ. . 1998 J. Leukoc. Biol. 63: 665–668.
Nicholson SE, Novak U, Ziegler SF and Layton JE. . 1995 Blood 86: 3698–3704.
Nicholson SE, Oates AC, Harpur AG, Ziemiecki A, Wilks AF and Layton JE. . 1994 Proc. Natl. Acad. Sci. USA 91: 2985–2988.
Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, Alexander WS, Metcalf D, Hilton DJ and Nicola NA. . 1999 EMBO J. 18: 375–385.
Nieborowska-Skorska M, Wasik MA, Slupianek A, Salomoni P, Kitamura T, Calabretta B and Skorski T. . 1999 J. Exp. Med. 189: 1229–1242.
Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, McBride OW and Leonard WJ. . 1993 Cell 73: 147–157.
Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Doherty PC, Grosveld GC and Ihle JN. . 1995 Science 270: 800–802.
Novick D, Cohen B and Rubinstein M. . 1994 Cell 77: 391–400.
O'Shea JJ, Notarangelo LD, Johnston JA and Candotti F. . 1997 J. Clin. Immunol. 17: 431–447.
Oh H, Fujio Y, Kunisada K, Hirota H, Matsui H, Kishimoto T and Yamauchi-Takihara K. . 1998 J. Biol. Chem. 273: 9703–9710.
Packham G, White EL, Eischen CM, Yang H, Parganas E, Ihle JN, Grillot DA, Zambetti GP, Nunez G and Cleveland JL. . 1998 Genes Dev. 12: 2475–2487.
Pallen CJ, Tan YH and Guy GR. . 1992 Curr. Opin. Cell. Biol. 4: 1000–1007.
Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G and Ihle JN. . 1998 Cell 93: 385–395.
Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, Miyake K, Nakauchi H, Shirasawa T and Saito T. . 1995 Immunity 3: 771–782.
Pawson T. . 1995 Nature 373: 573–580.
Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, Monpoux F, Van Rompaey L, Baens M, Van den Berghe H and Marynen P. . 1997 Blood 90: 2535–2540.
Pellegrini S and Dusanter-Fourt I. . 1997 Eur. J. Biochem. 48: 615–633.
Pellegrini M and Strasser A. . 1999 J. Clin. Immunol. 19: 365–377.
Pellegrini S, John J, Shearer M, Kerr IM and Stark GR. . 1989 Mol. Cell. Biol. 9: 4605–4612.
Pierce JH. . 1989 Biochim. Biophys. Acta 989: 179–208.
Pierce J, Potter M, Scott A, Humphries J, Aaronson SA and Ihle JN. . 1985 Cell 41: 685–692.
Pritchard MA, Baker E, Callen DF, Sutherland GR and Wilks AF. . 1992 Mamm. Genome 3: 36–38.
Quelle FW, Sato N, Witthuhn BA, Inhorn RC, Eder M, Miyajima A, Griffin JD and Ihle JN. . 1994 Mol. Cell. Biol. 14: 4335–4341.
Quelle FW, Thierfelder W, Witthuhn BA, Tang B, Cohen S and Ihle JN. . 1995 J. Biol. Chem. 270: 20775–20780.
Quelle FW, Wang J, Feng J, Wang D, Cleveland JL, Ihle JN and Zambetti GP. . 1998 Genes Dev. 12: 1099–1071.
Rane SG and Reddy EP. . 1994 Oncogene 9: 2415–2423.
Reddy EP, Smith MJ and Srinivasan A. . 1983 Proc. Natl. Acad. Sci. USA 80: 3623–3627.
Reddy EP, Korapati A, Chaturvedi P and Rane S. . 2000 Oncogene 19: 2532–2547.
Reed JC. . 1997 Nature 387: 773–776.
Riedy MC, Dutra AS, Blake TB, Modi W, Lal BK, Davis J, Bosse A, O'Shea JJ and Johnston JA. . 1996 Genomics 37: 57–61.
Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan KC, Yin L, Pennica D, Johnson Jr EM and Schreiber RD. . 1998 Cell 93: 373–383.
Rovera G, Valtieri M, Mavilio F and Reddy EP. . 1987 Oncogene 1: 29–35.
Rui H, Hirken RA and Farrar WL. . 1994 J. Biol. Chem. 269: 5364–5368.
Russell SM, Johnston JA, Noguchi M, Kawamura M, Bacon CM, Friedmann M, Berg M, McVicar DW, Witthuhn BA, Silvennoinen O, Goldman AS, Schmalstieg FC, Ihle JN, O'Shea JJ and Leonard W. . 1994 Science 266: 1042–1045.
Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, Migone TS, Noguchi M, Markert ML, Buckley RH, O'Shea JJ and Leonard W. . 1995 Science 270: 797–800.
Saharinen P, Takaluoma K and Silvennoinen O. . 2000 Mol. Cell. Biol. 20: 3387–3395.
Sakai I, Nabell L and Kraft AS. . 1995 J. Biol. Chem. 270: 18420–18427.
Sakai I and Kraft AS. . 1997 J. Biol. Chem. 272: 12350–12358.
Sakatsume M, Stancato LF, David M, Silvennoinen O, Saharinen P, Pierce J, Larner AC and Finbloom DS. . 1998 J. Biol. Chem. 273: 3021–3026.
Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I and Yoshimura A. . 2000 J. Biol. Chem. 875: 29293–29338
Schafer TS, Sanders LK and Nathans D. . 1995 Proc. Natl. Acad. Sci. USA 92: 9097–9101.
Schindler C. . 1999 Exp. Cell. Res. 253: 7–14.
Schindler C and Darnell Jr JE. . 1995 Annu. Rev. Biochem. 64: 621–651.
Shimoda K, Iwasaki H, Okamura S, Ohno Y, Kubota A, Arima F, Otsuka T and Niho Y. . 1994 Biochem. Biophys. Res. Commun. 203: 922–928.
Shuai K, Stark GR, Kerr IM and Darnell Jr JE. . 1993 Science 261: 1744–1746.
Silvennoinen O, Ihle JN, Schlessinger J and Levy DE. . 1993a Nature 366: 583–585.
Silvennoinen O, Witthuhn BA, Quelle FW, Cleveland JL, Yi T and Ihle JN. . 1993b Proc. Natl. Acad. Sci. USA 90: 8429–8433.
Smith A, Metcalf D and Nicola NA. . 1997 EMBO J. 16: 451–464.
Soh J, Donnelly RJ, Kotenko S, Mariano TM, Cook JR, Wang N, Emanuel S, Schwartz B, Miki T and Pestka S. . 1994 Cell 76: 793–802.
Stahl N, Boulton TG, Farruggella T, Ip NY, Davis S, Witthuhn BA, Quelle FW, Silvennoinen O, Barbieri G, Pellegrini S, Ihle JN and Yancopoulos GD. . 1994 Science 263: 92–95.
Stancato LF, Sakatsume M, David M, Dent P, Dong F, Petricoin EF, Krolewski JJ, Silvennoinen O, Saharinen P, Pierce J, Marshall CJ, Sturgill T, Finbloom DS and Larner AC. . 1997 Mol. Cell. Biol. 17: 3833–3840.
Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA and Hilton DJ. . 1997 Nature 387: 917–921.
Strous GJ, van Kerkhof P, Govers R, Ciechanover A and Schwartz AL. . 1996 EMBO J. 15: 3806–3812.
Takahashi T and Shirasawa T. . 1994 FEBS Lett. 342: 124–128.
Takaki S, Kanazawa H, Shiiba M and Takatsu K. . 1994 Mol. Cell. Biol. 14: 7404–7413.
Tanaka N, Asao H, Ohbo K, Ishii N, Takeshita T, Nakamura M, Sasaki H and Sugamura K. . 1994 Proc. Natl. Acad. Sci. USA 91: 7272–7275.
Tanner JW, Chen W, Young RL, Longmore GD and Shaw AS. . 1995 J. Biol. Chem. 270: 6523–6530.
Tanuma N, Nakamura K, Shima H and Kikuchi K. . 2000 J. Biol. Chem. 275: 28216–28221.
Thomis DC, Gurniak CB, Tivol E, Sharpe AH and Berg LJ. . 1995 Science 270: 794–797.
Turkson J, Bowman T, Adnane J, Zhang Y, Djeu YJ, Sekharam M, Frank DA, Holzman LB, Wu J, Sebti S and Jove R. . 1999 Mol. Cell. Biol. 19: 7519–7528.
Uchida K, Yoshimura A, Inazawa J, Yanagisawa K, Osada H, Masuda A, Saito T, Takahashi T and Miyajima A. . 1997 Cytogenet. Cell. Genet. 78: 209–212.
Valtieri M, Tweardy DJ, Caracciolo D, Johnson K, Mavilio F, Altmann S, Santoli D and Rovera G. . 1987 J. Immunol. 138: 3829–3835.
VanderKuur JA, Wang X, Zhang L, Campbell GS, Allevato G, Billestrup N, Norstedt G and Carter-Su C. . 1994 J. Biol. Chem. 269: 21709–21717.
Velazquez L, Fellous M, Stark GR and Pellegrini S. . 1992 Cell 70: 313–322.
Velazquez L, Mogensen KE, Barbieri G, Fellous M, Uze G and Pellegrini S. . 1995 J. Biol. Chem. 270: 3327–3334.
Verbsky JW, Bach EA, Fang YF, Yang L, Randolph DA and Fields LE. . 1996 J. Biol. Chem. 271: 13976–13980.
Verdier F, Chretien S, Muller O, Varlet P, Yoshimura A, Gisselbrecht S, Lacombe C and Mayeux P. . 1998 J. Biol. Chem. 273: 28185–28190.
Wang Y, Morella KK, Ripperger J, Lai CF, Gearing DP, Fey GH, Campos SP and Baumann H. . 1995 Blood 86: 1671–1679.
Ward AC, Touw I and Yoshimura A. . 2000 Blood 95: 19–29.
Watling D, Guschin D, Muller M, Silvennoinen O, Witthuhn BA, Quelle FW, Rogers NC, Schindler C, Stark GR, Ihle JN and Kerr IM. . 1993 Nature 366: 166–170.
Watowich SS, Hilton DJ and Lodish HF. . 1994 Mol. Cell. Biol. 14: 3535–3549.
Weiss A and Schlessinger J. . 1998 Cell 94: 277–280.
Wilks AF. . 1989 Proc. Natl. Acad. Sci. USA 86: 1603–1607.
Wilks AF. . 1991 Methods Enzymol. 200: 533–546.
Winston LA and Hunter T. . 1995 J. Biol. Chem. 270: 30837–30840.
Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O and Ihle JN. . 1993 Cell 74: 227–236.
Witthuhn BA, Silvennoinen O, Miura O, Lai KS, Cwik C, Liu ET and Ihle JN. . 1994 Nature 370: 153–157.
Xu X, Sun YL and Hoey T. . 1996 Science 272: 794–797.
Yamamoto K, Quelle FW, Thierfelder WE, Kreider BL, Gilbert DJ, Jenkins NA, Copeland NG, Silvennoinen O and Ihle JN. . 1994 Mol. Cell. Biol. 14: 4342–4349.
Yamauchi T, Kaburagi Y, Ueki K, Tsuji Y, Stark GR, Kerr IM, Tsushima T, Akanuma Y, Komuro I, Tobe K, Yazaki Y and Kadowaki T. . 1998 J. Biol. Chem. 273: 15719–15726.
Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN and Yoshimura A. . 1999 EMBO J. 18: 1309–1320.
Yasukawa H, Sasaki A and Yoshimura A. . 2000 Annu. Rev. Immunol. 18: 143–164.
Yetter D. . 1995 Nurs. Manage. 26: 60.
Yin T, Shen R, Feng GS and Yang YC. . 1997 J. Biol. Chem. 272: 1032–1037.
Yin T, Tsang ML and Yang YC. . 1994 J. Biol. Chem. 269: 26614–26617.
Yoshimura A. . 1998a Cytokine Growth Factor Rev. 9: 197–204.
Yoshimura A. . 1998b Leukemia 12: 1851–1857.
Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, Hara T and Miyajima A. . 1995 EMBO J. 14: 2816–2826.
You M, Yu DH and Feng GS. . 1999 Mol. Cell. Biol. 19: 2416–2424.
Yu CL and Burakoff SJ. . 1997 J. Biol. Chem. 272: 14017–14020.
Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J and Jove R. . 1995 Science 269: 81–83.
Zeng YX, Takahashi H, Shibata M and Hirokawa K. . 1994 FEBS Lett. 353: 289–293.
Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, Kile BJ, Kent SB, Alexander WS, Metcalf D, Hilton DJ, Nicola NA and Baca M. . 1999 Proc. Natl. Acad. Sci. USA 96: 2071–2076.
Zhao Y, Wagner F, Frank SJ and Kraft AS. . 1995 J. Biol. Chem. 270: 13814–13818.
Zhao YJ, Hanson EP, Chen YQ, Magnuson K, Chen M, Swann PG, Wange RL, Changelian PS and O'Shea JJ. . 1997 Proc. Natl. Acad. Sci. USA 94: 13850–13855.
Acknowledgements
This work was supported by grants from the NIH (ES09225-02 and CA68239-04).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Rane, S., Reddy, E. Janus kinases: components of multiple signaling pathways. Oncogene 19, 5662–5679 (2000). https://doi.org/10.1038/sj.onc.1203925
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1203925
Keywords
This article is cited by
-
Carcinogenicity of nicotine and signal pathways in cancer progression: a review
Environmental Chemistry Letters (2024)
-
Janus Kinase 3 phosphorylation and the JAK/STAT pathway are positively modulated by follicle-stimulating hormone (FSH) in bovine granulosa cells
BMC Molecular and Cell Biology (2023)
-
The pan-JAK inhibitor LAS194046 reduces neutrophil activation from severe asthma and COPD patients in vitro
Scientific Reports (2022)
-
Role of intracellular signaling pathways and their inhibitors in the treatment of inflammation
Inflammopharmacology (2021)
-
STAT3 transcription factor as target for anti-cancer therapy
Pharmacological Reports (2020)
