
Abstract
A systematic search was made for uniparental disomy in carriers of apparently balanced chromosome translocations who also had unexplained abnormalities of mental or physical development. Of 65 families studied, biparental origin of both translocated chromosomes was demonstrated in 64, and only 1 case of maternal uniparental disomy of chromosome 14 was detected in the carrier of a Robertsonian t(13q14q). We conclude that uniparental disomy is a rare occurrence in this population.
Similar content being viewed by others

Introduction
The term uniparental disomy was originally used by Eric Engel [1] in 1980. Under normal circumstances humans have 22 pairs of autosomes and 1 pair of sex chromosomes, one of each homologous pair originating from the father and one from the mother. In uniparental disomy, diploid offspring inherit both homologues of a chromosome pair from 1 parent. Unless there are obvious cytogenetic heteromorphisms to distinguish the parental origin of the homologues, uniparental disomy is cytogenetically undetectable.
There are a number of proposed mechanisms by which uniparental disomy might arise. Nondisjunctional events during meiosis may result in aneuploid gametes which, on fertilization, give rise to uniparental disomy in the zygote by one of three mechanisms:
-
(1)
Gamete complementation: Engel’s original suggestion was that fusion of a nullisomic gamete with a gamete disomic for the same chromosome would result in a diploid zygote.
-
(2)
Monosomy duplication: A zygote resulting from the fusion of a monosomic gamete with a nullisomic gamete would be nonviable unless the single chromosome duplicated itself, resulting in isodisomy of that chromosome.
-
(3)
Trisomy rescue: When a disomic gamete fuses with a normal monosomic gamete the resulting zygote is trisomie. If the zygote corrects the initial trisomy by loss of the supernumerary chromosome, random loss would result in a normal zygote in 2/3 of cases, and uniparental disomy in 1/3.
Postzygotic mitotic nondisjunction may also result in uniparental disomy, if one homologue of a chromosome pair is lost and the other duplicated to retain euploidy. Mitotic events would result in mosaicism for uniparental disomy unless they occur very early on in embryogenesis.
Uniparental disomy may affect development if an autosomal recessive gene mutation is unmasked by autozygosity, or if the chromosome involved bears regions subject to imprinting. Imprinting is defined as the differential expression of a gene dependent upon the parent of origin. Studies in mice have demonstrated that uniparental disomy of certain chromosomes appears to have no phenotypic effect, presumably because they do not bear imprinted regions. However, some chromosomes demonstrate different phenotypic effects dependent upon the parent of origin, while uniparental disomy of others appears to be nonviable [2–4]. Situations analogous to those caused by imprinting in mice are also evident in humans, indicating that similar imprinting mechanisms exist, and from mouse studies it would be predicted that imprinted regions are present on human chromosomes 2, 5, 6, 7, 9, 11, 15, 16, 19, 20, 21 and 22 [4, 5]. Thus uniparental disomy can provide a tool for the determination of both imprinted regions in the human genome and the mapping of autosomal recessive genes.
Relatively few cases of uniparental disomy in humans have yet been described in the literature (for summary, see table 1). This could be a true reflection of their rarity, or result from the lack of a simple screening test. In some patients the clinical features associated with uniparental disomy are clearly due to an autosomal recessive disease such as cystic fibrosis [6, 7] or rod monochromacy [8]. In others, the clinical features common to all patients with uniparental disomy of the same chromosome may result from the presence of imprinted regions on the chromosome. Three cases of maternal uniparental disomy of chromosome 7 have been described, and short stature is a feature in each case [6, 7, 9]. Maternal uniparental disomy of chromosome 16 has now been described on seven occasions [10–13] and may result in intrauterine growth retardation. However, each case has been associated with a trisomie cell line for chromosome 16 which may have been the cause of the poor placental function. Uniparental disomy for chromosomes 6 (paternal), 21 (both maternal and paternal), 22 (maternal) and XY (paternal) have all been reported to be associated with normal development [14–19].
Few syndromes associated with imprinted regions are well recognized. An exception is uniparental disomy of chromosome 15 which, when it is maternal, results in Prader-Willi syndrome and, when paternal, in Angelman syndrome. Another well-characterized condition in which imprinting is known to play a part is Beckwith-Wiedemann syndrome. Henry et al. [20] have demonstrated that 20% of sporadic cases of Beckwith-Wiedemann syndrome have paternal uniparental isodisomy (autozygosity) for 11p15.5. In some cases the loss of maternal alleles was shown to be a mitotic event as evidenced by somatic mosaicism for the uniparental disomy.
It has been shown in Prader-Willi syndrome that the maternal age is greater in patients with maternal uniparental disomy compared with those with a paternal deletion of 15q12–13 [21, 22]. This is consistent with the fact that the incidence of oocyte aneuploidy increases with maternal age, and supports the three mechanisms proposed for uniparental disomy. It would be anticipated that gamete complementation is the least likely mechanism, since errors in both maternal and paternal meiosis are required. Evidence of trisomy 15 in chorionic villus samples from 2 Prader-Willi patients indicates that trisomy correction is the mechanism by which they have arisen. Table 1 shows that in a total of 30 patients, excluding those ascertained for Prader-Willi, Angelman and Beckwith-Wiedemann syndromes, maternal uniparental disomy (21 cases) is more common than paternal (9 cases).
In order to gain further insight into uniparental disomy in humans, we have studied a population which we reasoned should be at increased risk of uniparental disomy. Since any situation that predisposes to the production of aneuploid gametes would be expected to predispose to uniparental disomy, our systematic search began with the selection of a population of individuals who should be at increased risk of uniparental disomy because their parents were at increased risk of meiotic nondisjunction.
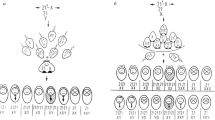
Carriers of Robertsonian and certain reciprocal translocations have been identified as a high risk group, since the incidence of nondisjunction is increased for those chromosomes involved in the translocation. Under normal circumstances, during meiosis I, homologous chromosomes pair and recombination occurs before the homologues separate and move to the two daughter cells. In cells with structural rearrangements of the chromosomes the homologues are unable to pair in the usual way (fig. 1).

Mechanisms resulting in uniparental disomy in carriers of (A) Robertsonian and (B) reciprocal translocations. 1. At meiosis, (a) adjacent segregation and (b) 3:1 segregation. 2. Fusion of gamete disomic for the black chromosome with a normal monosomic gamete (hatched chromosome). 3. Zygote trisomie for the black/hatched chromosome. 4. Trisomy correction results in either (a) a normal zygote or (b) uniparental disomy of the black chromosome.
In Robertsonian heterozygotes the translocation chromosome and the two normal homologues usually synapse as a trivalent. Alternate segregation results in balanced/normal gametes. Adjacent segregation results in disomic and nullisomic gametes [23]. In reciprocal translocations the two translocation and the two normal homologues form a quadrivalent at pachytene. They can then segregate in a number of ways. Alternate segregation results in normal/balanced daughter cells, adjacent 2:2 segregation results in unbalanced daughter cells and adjacent 3:1 segregation can lead to aneuploid daughter cells. Recent studies using fluorescence in situ hybridization on human meiotic preparations have demonstrated that the segregation patterns in carriers of reciprocal translocations vary between individuals, and that the presence of interstitial chiasmata can influence the products of meiosis [24, 25]. A number of studies have demonstrated that carriers of Robertsonian and reciprocal translocations are at increased risk of producing unbalanced offspring [e.g. 26, 27].
A beneficial feature of searching for uniparental disomy in carriers of a translocation is that the chromosomes at risk of being uniparentally disomic can be targeted since they are those involved in the translocation. Thus, for each proband, only two chromosomes need to be studied, whereas it is difficult to screen for uniparental disomy in the general population since all the chromosomes must be tested for parental origin. Our postulate that translocation carriers are at increased risk of uniparental disomy is born out by a search of the literature which reveals that there have been 14 cases of uniparental disomy associated with translocations. Of the 14, 8 are associated with homologous translocations, 4 with nonhomologous Robertsonian translocations and 2 with reciprocal translocations. In addition, there are 2 cases associated with isochromosomes (table 1).
The Study Population
Individuals were identified initially from the Wes-sex Regional Genetics Laboratory. A systematic search was made for those who presented with an unexplained abnormality of mental or physical development including infertility and who were subsequently found to have an apparently balanced Robertsonian or reciprocal translocation. The clinical criteria for inclusion were wide, as, for the majority of chromosomes, it is not known how, or if, uniparental disomy affects development.
All families were contacted via a letter to the physician who initially referred the patient for chromosome analysis. An attempt was made to contact all families although, as our records went back 15 years, it was often impossible to trace the patients. In those families who agreed to participate, 10–20 ml of blood was obtained from the proband and both parents. As we had no idea what proportion would show uniparental disomy, a thorough clinical examination was only proposed for those subsequently found to have uniparental disomy.
Following this initial approach, collaboration with a second Regional Genetics Unit, North West Thames Health Authority, was initiated and an identical search was made. Clinical geneticists in the UK and Europe were also invited to collaborate.
Methods
Cytogenetic A nalysis
Chromosome analysis was carried out on peripheral blood lymphocytes from each proband and both parents where possible. Translocation breakpoints were determined after G-banding of metaphase chromosomes [28].
Molecular Techniques
DNA was extracted from whole blood by a salt precipitation technique [29]. Parental origin of the chromosomes involved in the translocations was determined using two molecular techniques: (1) analysis by standard Southern blot methods and hybridization with radioactive probes [30, 31] and (2) amplification of polymorphic microsatellite repeat sequences [32, 33] using standard PCR amplification [34]. PCR products were visualized using a 6% denaturing Polyacrylamide gel followed by autoradiography. Details of the probes and primers used may be obtained on request from the authors.
Since we were searching for uniparental disomy of whole chromosomes, a single biparental result, or two results, one demonstrating a maternal contribution and one demonstrating a paternal contribution, was sufficient to exclude uniparental disomy for each chromosome.
Results
A total of 94 families were contacted via the Wessex Genetics Service, and 22 families via the North West Thames Genetics Service. To date we have tested 44 complete families (proband and both parents) and 12 partial families (proband and 1 parent). We have also tested 9 further families supplied by other centres. In total, a search for uniparental disomy was made in 65 probands. Details of each case are given in tables 2 and 3.
Our study population represented a total of 130 target chromosomes and uniparental disomy was excluded for 129 of these, i.e. biparental origin was determined for both translocated chromosomes in 64 probands. Only a single case of uniparental disomy was detected (family No. Z57). The proband was the carrier of a Robertsonian t(13; 14)mat and was found to have maternal uniparental disomy of chromosome 14. This patient was one of the first three studied and full details have been published [35].
Discussion
The main conclusion that can be drawn from this study is that uniparental disomy is a relatively rare event among balanced translocation carriers, when they have been ascertained because of clinical abnormality. In this series of 65 cases, uniparental disomy was found only in patient Z57, and its relationship to his clinical condition is not yet clear (see below).
Of the 72 cases of uniparental disomy reported in the literature and included in table 1, 14 were associated with a translocation showing that uniparental disomy does occur in balanced chromosome translocations. Only 2 of the 14 were associated with reciprocal translocations, of which 1 involved chromosomes unrelated to the uniparental disomy, suggesting that uniparental disomy may well be rare in carriers of reciprocal translocations. However, prior to this study the incidence of uniparental disomy in translocation carriers was unknown.
The only significant series of patients with uniparental disomy ascertained for clinical reasons are those with Prader-Willi syndrome where uniparental disomy accounts for 15–20% of cases. It is of interest that from two studies investigating a total of 25 cases of maternal uniparental disomy of chromosome 15, one was associated with a t(13; 15) Robertsonian translocation, and one with a reciprocal translocation involving nonrelated chromosomes (t(8;18)) [21,22].
The association between clinical anomalies and balanced chromosomal rearrangemerits is documented but contentious [36]. There is, however, generally considered to be an empiric risk of 5–10% for a developmental anomaly in de novo reciprocal balanced translocation carriers when detected prenatally [23]. We would suggest that few of these developmental anomalies are caused by uniparental disomy, and, on the basis of our results, the search for uniparental disomy in translocation carriers is unlikely to be very productive. An exception may be the rare examples of balanced translocations involving homologous chromosomes, particularly where the chromosome involved has a high probability of having an imprinted region [37].
Since the publication of the 1 case of maternal uniparental disomy of chromosome 14 detected in our study [35], 2 other cases have been described [8,38]. There are clinical similarities among these patients which might help in the recognition of further cases (table 4). Affected individuals had short stature, scoliosis, hydrocephalus and premature puberty. It is of interest that the patient described by Antonarakis et al. [38] was shown to have a trisomy 14 cell line in 5% of blood lymphocytes. Features described in mosaic trisomy 14 [39] show similarities to the 3 cases of maternal uniparental disomy of 14 (table 4) and it could be that it is mosaicism for trisomy 14 that accounts for the clinical picture. From mouse studies it is not yet clear whether imprinted regions would be predicted on human chromosome 14 [5].
This is the first study which has searched systematically for uniparental disomy for chromosomes other than for chromosome 15 in Prader-Willi and Angelman syndromes, and chromosome 11 in Beckwith-Wiedemann syndrome. Uniparental disomy for some chromosomes may be lethal, while for other chromosomes, for example paternal chromosome 6, chromosome 21 and maternal chromosome 22, uniparental disomy is associated with a normal phenotype. Our study was designed to answer the question of the incidence of uniparental disomy in phenotypically abnormal carriers of apparently balanced translocations, and does not provide information on the total incidence of uniparental disomy in translocation carriers. We did not detect many cases of uniparental disomy, thus showing that uniparental disomy is not a common cause of phenotypic abnormality in this population.
References
Engel E: A new genetic concept: Uniparental disomy and its potential effect, isodisomy. Am J Med Genet 1980;6:137–143
Searle AG, Beechey CV: Noncomplementation phenomena and their bearing on nondisjunctional effects; in Dellarcho VL, Voytek PE, Hollaender A (eds): Aneuploidy: Etiology and Mechanisms. New York, Plenum Press, 1985, pp 363–376.
Cattanach BM, Kirk M: Differential activity of maternally and paternally derived chromosome regions in mice. Nature 1985;315:496–498
Hall JG: Genomic imprinting: Review and relevance to human diseases. Am J Hum Genet 1990;46:857–873
Searle AG, Peters J, Lyon MF, Hall JG, Evans EP, Edwards JH, Buckle VJ: Chromosome maps of man and mouse. IV. Ann Hum Genet 1989;53:89–140
Spence JE, Perciaccante RG, Greig GM, Willard HF, Ledbetter DH, Hejtmancik JF, Pollack MS, O’Brien WE, Beaudet AL: Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet 1988;42:217–226
Voss R, Ben-Simon E, Avital A, Godfrey S, Zlotogora J, Dagan J, Tikochinski Y, Hillel J: Isodisomy of chromosome 7 in a patient with cystic fibrosis: Could uniparental disomy be common in humans? Am J Hum Genet 1989;45:373–380
Pentao L, Lewis RA, Ledbetter DH, Patel PI, Lupski JR: Maternal uniparental isodisomy of chromosome 14. Association with autosomal recessive rod monochromacy. Am J Hum Genet 1992;50:690–699
Spotila LD, Sereda L, Prockop DJ: Partial isodisomy for maternal chromosome 7 and short stature in an individual with a mutation at the COLIA2 locus. Am J Hum Genet 1992;51:1396–1405
Dwomiczak B, Koppers B, Kurlemann G, Holzgreve W, Horst J, Miny P: Uniparental disomy with normal phenotype. Lancet 1992;340:1285.
Bennett P, Vaughan J, Henderson D, Loughna S, Moore G: Association between confined placental trisomy, fetal uniparental disomy, and early intrauterine growth retardation. Lancet 1992;340:1284–1285
Kalousek DK, Langlois S, Barrett I, Yam I, Wilson DR, Howard-Peebles PN, Johnson MP, Giorgiutti E: Uniparental disomy for chromosome 16 in humans. Am J Hum Genet 1993;52:8–16
Sutcliffe M J, Mueller OT, Gallardo LA, Papenhausen PR, Tedesco TA: Maternal isodisomy in a normal 46,XX following trisomie conception. Am J Hum Genet 1993;53(suppl):1464.
Welch TR, Beischel LS, Choi E, Balakrishnan K, Bishof NA: Uniparental isodisomy 6 associated with deficiency of the fourth component of complement. J Clin Invest 1990;86:675–678
Créau-Goldberg N, Gegonne A, Delabar J, Cochet C, Cabanis M-O, Stehelin D, Turleau C, de Grouchy J: Maternal origin of a de novo balanced t(21q21q) identified by ets-2 polymorphism. Hum Genet 1987;76:396–398
Blouin J-L, Avramopoulos D, Pangalos C, Antonarakis SE: Normal phenotype with paternal uniparental isodisomy for chromosome 21. Am J Hum Genet 1993;53(suppl): 1129.
Palmer CG, Schwartz S, Hodes ME: Transmission of a balanced homologous t(22q;22q) translocation from mother to normal daugther. Clin Genet 1980;17:418–422
Kirkels VGHJ, Hustinx TWJ, Scheres JMJC: Habitual abortion and translocation (22q;22q): Unexpected transmission from a mother to her phenotypically normal daughter. Clin Genet 1980;18:456–461
Vidaud D, Vidaud M, Plassa F, Gazengel C, Noel B, Goossens M: Father-to-son transmission of haemophilia A due to uniparental disomy. Am J Hum Genet 1989;45(suppl A226):889.
Henry I, Puech A, Riesewijk A, Ahnine L, Mannens M, Beldjord C, Bitoun P, Tournade MF, Landrieu P, Junien C: Somatic mosaicism for partial paternal isodisomy in Wiedemann-Beckwith syndrome: A postfertilization event. Eur J Hum Genet 1993;1:19–29
Robinson WP, Bottani A, Yagang X, Balakrishman J, Binkert F, Mächler M, Prader A, Schinzel A: Molecular, cytogenetic, and clinical investigations of Prader-Willi syndrome patients. Am J Hum Genet 1991;49:1219–1234
Mascari MJ, Gottlieb W, Rogan PK, Butler MG, Waller DA, Armour JAL, Jeffreys AJ, Ladda RL, Nicholls RD: The frequency of uniparental disomy in Prader-Willi syndrome: Implications for molecular diagnosis. N Engl J Med 1992;326:1599–1607
Gardner RJM, Sutherland GR: Chromosome Abnormalities and Genetic Counselling. Oxford, Oxford University Press, 1989.
Goldman ASH, Hultén MA: Meiotic analysis by FISH of a human male 46, XY, t(15;20)(q11.2;q11.2) translocation heterozygote: Quadrivalent configuration, orientation and first meiotic segregation. Chromosoma 1993;102:102–111
Goldman ASH, Hultén MA: Analysis of chiasma frequency and first meiotic segregation in a human male reciprocal translocation heterozygote, t(1;11)(p36.3;q13.1) using fluorescence in situ hybridisation. Cytogenet Cell Genet 1993,63: 16–23.
Jacobs PA, Frackiewicz A, Law P, Hilditch CJ, Morton NE: The effect of structural aberrations of the chromosomes on reproductive fitness in man. II. Results. Clin Genet 1975;8:169–178
Boué A, Gallano P: A collaborative study of the segregation of inherited chromosome structural rearrangements in 1,356 prenatal diagnoses. Prenat Diagn 1984;4:45–67
Seabnght M: A rapid banding technique for human chromosomes. Lancet 1971;ii:971–972
Miller SA, Dykes DD, Polesky HF: A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16:1215.
Feinberg AP, Vogelstein B: A technique for radiolabelling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 1983;132:6–13
Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. New York, Cold Spring Harbor Laboratory Press, 1989.
Weber JL, May PE: Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet 1989;44:388–396
Edwards A, Civitello A, Hammond HA, Caskey CT: DNA typing and genetic mapping with trimeric and tetrameric tandem repeats. Am J Hum Genet 1991;49:746–756
Hudson TJ, Engelstein M, Lee MK, Ho EC, Rubenfield MJ, Adams CP, Housman DE, Dracopoli NC: Isolation and chromosomal assignment of 100 highly informative human simple sequence repeat polymorphisms. Genomics 1992;13:622–629
Temple IK, Cockwell A, Hassold T, Pettay D, Jacobs P: Maternal uniparental disomy for chromosome 14. J Med Genet 1991;28:511–514
Fryns JP, Kleczkowska A, Kubien E, Van den Berghe H: Excess of mental retardation and/or congenital malformation in reciprocal translocations in man. Hum Genet 1986;72:1–8
Freeman SB, May KM, Pettay D, Fernhoff PM, Hassold TJ: Paternal uniparental disomy in a child with a balanced 15; 15 translocation and Angelman syndrome. Am J Med Genet 1993;45:625–630
Antonarakis SE, Blouin JL, Maher J, Avramopoulos D, Thomas G, Talbot CC: Maternal uniparental disomy for human chromosome 14, due to loss of a chromosome 14 from somatic cells with t(13;14) trisomy 14. Am J Hum Genet 1993;52:1145–1152
Fujimoto A, Allanson J, Crowe CA, Lipson MH, Johnson VP: Natural history of mosaic trisomy 14 syndrome. Am J Med Genet 1992;44:189–196
Carpenter NJ, Say B, Barber ND: A homozygote for pericentric inversion of chromosome 4. J Med Genet 1982;19:469–471
Lindenbaum RH, Woods CG, Norbury CG, Povey S, Rysleckl G: An individual with maternal disomy of chromosome 4 and iso(4p), iso(4q). Am J Hum Genet 1991;49(suppl 285): 1582.
Schonberg SA, Eggerding FA, Norton ME, Cox VA, Chehab F, Epstein CJ: Growth retardation in a chromosomally balanced child with ‘isochromosomes’ of 7p and 7q: A possible manifestation of uniparental isodisomy. Am J Hum Genet 1992;51(supplA87):336.
Willatt LR, Davison BCC, Goudie D, Alexander J, Dyson HM, Jenks PE, Ferguson-Smith ME: A male with trisomy 9 mosaicism and maternal uniparental disomy for chromosome 9 in the euploid cell line. J Med Genet 1992;29:742–744
Kousseff BG, Gallardo LA, Mueller OT: Unusual clinical presentation associated with uniparental disomy of chromosome 10 in a child presymptomatic for multiple endocrine neoplasia type 2A. Am J Hum Genet 1992;51(suppl A219):863.
Grundy P, Telzerow P, Paterson MC, Haber D, Berman B, Li F, Garber J: Chromosome 11 uniparental isodisomy predisposing to embryonal neoplasms. Lancet 1991;338:1079–1080
Henry I, Bonaiti-Pellié C, Chehensse V, Beldjord C, Schwartz C, Utermann G, Junien C: Uniparental paternal disomy in a genetic cancerpredisposing syndrome. Nature 1991;351:665–667
Beldjord C, Henry I, Bennani C, Vanhaeke D, Labie Uniparental disomy: A novel mechanism for thalassemia major. Blood 1992;80:287–289.
Hassold T, Jacobs PA, Leppert M, Sheldon M: Cytogenetic and molecular studies of trisomy 13. J Med Genet 1987;24:725–732
Wang JCC, Passage MB, Yen PH, Shapiro LJ, Mohandas TK: Uniparental heterodisomy for chromosome 14 in a phenotypically abnormal familial balanced 13/14 Robertsonian translocation carrier. Am J Hum Genet 1991;48:1069–1074
Diamond TM, Mueller OT, Sutcliffe M, Papenhausen PR, Tedesco TA, Kousseff BG: Uniparental disomy for chromosome 14 — Evidence for an imprinting effect. Am J Hum Genet 1993;53(suppl):541.
Nicholls RD, Knoll JHM, Butler MG, Karam S, Lalande M: Genetic imprinting suggested by maternal heterodisomy in non-deletion Prader-Willi syndrome. Nature 1989;342:281–285
Hamabe J, Fukushima Y, Harada N, Abe K, Matsuo N, Nagai T, Ycshioka A, Tonoki H, Tsukino R, Niikawa N: Molecular study of the Prader-Willi syndrome: Deletion, RFLP, and phenotype analyses of 50 patients. Am J Med Genet 1991;41:54–63
Cassidy SB, Lai LW, Erickson RP, Magnuson L, Thomas E, Gendron R, Herrmann J: Trisomy 15 with loss of the paternal 15 as a cause of Prader-Willi syndrome due to maternal disomy. Am J Hum Genet 1992;51:701–708
Purvis-Smith SG, Saville T, Manass S, Yip MY, Lam-Po-Tang PRL, Duffy B, Johnston H, Leigh D, Mcdonald B: Uniparental disomy 15 resulting from correction of an initial trisomy 15. Am J Hum Genet 1992;50:1348–1350
Malcolm S, Clayton-Smith J, Nichols M, Robb S, Webb T, Armour JAL, Jeffreys AJ, Pembrey ME: Uniparental paternal disomy in Angelman’s syndrome. Lancet 1991;337:694–696
Nicholls RD, Pai GS, Gottlieb W, Cantú ES: Paternal uniparental disomy of chromosome 15 in a child with Angelman syndrome. Ann Neurol 1992;32:512–518
Smeets DFCM, Hamel BCJ, Nelen MR, Smeets HJM, Bollen JHM, Smits APT, Ropers HH, van Oost BA: Prader-Willi syndrome and Angelman syndrome in cousins from a family with a translocation between chromosome 6 and chromosome 15. N Engl J Med 1992;326:807–811
Ngo KY, Lee J, Dixon B, Liu D, Jones OW: Paternal uniparental isodisomy in a hydrops fetalis α-thalassemia fetus. Am J Hum Genet 1993;53(suppl): 1207
Petersen MB, Bartsch O, Adelsberger PA, Mikkelsen M, Schwinger E, Antonarakis SE: Uniparental isodisomy due to duplication of chromosome 21 occurring in somatic cells monosomic for chromosome 21. Genomics 1992;13:269–274
Schinzel AA, Robinson WP, Binkert F, Torresani T, Werder EA: Exclusively paternal X chromosomes in a girl with short stature. Hum Genet 1993;92:175–178
Acknowledgements
The work of R.S. James is supported by a grant from the Wellcome Trust. We would like to thank those clinicians who supplied us with DNA samples: J. Goodship, M. Ramos, J. Tolmie, A. Wilkie, A. Fryer, M. Jonsweijer, G. Lewis and P. Turnpenny. We are also grateful to our colleagues in the Wessex Regional Genetics Laboratory, notably John Harvey and Julia Fisher in the Molecular Genetics Laboratory for their helpful support, and colleagues in the Cytogenetics Laboratory for undertaking chromosome analysis. We also thank Annette Dash wood for her help with the manuscript.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
James, R.S., Temple, I.K., Patch, C. et al. A Systematic Search for Uniparental Disomy in Carriers of Chromosome Translocations. Eur J Hum Genet 2, 83–95 (1994). https://doi.org/10.1159/000472348
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1159/000472348
Key Words
This article is cited by
-
The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation
Nature Genetics (1995)
