
Abstract
Linkage analysis was carried out in two British families with incontinentia pigmenti (IP). Both showed exclusion at several markers in Xp and proximal Xq and showed probable linkage to the DXS52 and F8C loci in Xq28. This suggests that in these families the disease locus is IP2. Using a method based on the androgen receptor gene, and confirming the results where possible at the PGK-1 and DXS255 loci, it was shown that in affected females the maternally inherited X chromosome, where it could be identified, is inactive in the majority of cells.
Similar content being viewed by others

Introduction
Incontinentia pigmenti (IP, MIM 308300) is a rare X-linked genodermatosis characterized by disturbance of skin pigmentation with associated hair, nail and teeth anomalies [1]. It is sometimes associated with neurological and eye defects. Erythematous and vesicular skin at birth may develop into verrucous lesions which are later replaced by linear or blotchy areas of increased or decreased pigmentation following Blaschko’s lines. IP is found predominantly in females, with male patients being rare [2] and possibly the result of mosaicism or X chromosome abnormalities [3].
A proportion of IP cases are sporadic. Several of these have been shown to be associated with X;autosome translocations with breakpoints in Xp11 [4]. These are considered to disrupt a locus designated IP1. Familial cases of IP show sex-linked, dominant inheritance with lethality in hemizygous males. Family studies reported by several groups have excluded various regions of the X chromosome [5–7] and shown linkage to markers at Xq28 [8]. This locus has been designated IP2. Multipoint linkage analysis with nine terminal long arm markers and consideration of families with particular recombinants [9] suggested a location of IP2 distal to F8C in Xq28. The observed heterogeneity of translocation breakpoints at Xp11 [4] and a report of families showing no linkage to either IP1 or IP2 [10] may indicate the existence of other loci.
The phenotype of affected females has been interpreted on the basis that lesions are caused by the death of cells which have the mutated gene on the active X chromosome being replaced by cells where the normal gene is active [11]. Selection against the expression of the mutant gene has been confirmed in skin fibroblast cultures and also to varying degrees in hematopoietic tissues [12].
We report linkage data in one family with 7 clearly affected females which suggests Xq28 as the probable gene location (IP2 locus). DNA prepared from peripheral blood lymphocytes (PBL) in the affected individuals of this family, and in a second small pedigree, demonstrate striking, nonrandom X chromosome activity.
Materials and Methods
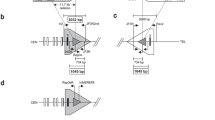
The pedigrees of the two families are given in figure 1. In family 1, the 2 carrier females in generation I have minimal or no clinical evidence of IP. I.3 (aged 60) has a convincing history of the typical rash soon after birth and some ‘odd’ birthmarks in childhood which have since faded. I.2 has no hard clinical evidence of IP although she had a dental clearance at the age of 20 and admits that her teeth were misshapen and similar to those of her affected daughters. She also had two miscarriages at 3 and 4 months with no definable fetus. All the gene carriers in the second generation have either a convincing history or convincing signs or both. The clinical signs include hyper- and hypopigmentation respecting Blaschko’s lines, patchy alopecia and typical dental dysplasia. II.5 has retinal dysplasia and has had a retinal detachment leaving her partially sighted. II.1, II.3 and II.6 have no features of IP and are considered to be unaffected. II.1 has 1 male child, II.6 has 2 female children and neither have a history of miscarriage. All members of this pedigree have normal intelligence and there is no history of seizures.

Pedigrees of the two IP families with the individuals studied numbered as in the text. Genotypes at the three loci used to investigate X chromosome activity are given in the order: DXS255, AR and PGK-1. Alleles are separated with | where phase is known and: where phase has been assumed, with the paternal allele on the left-hand side in each case. Where phase is unknown a space separates the alleles. Asterisks mark alleles shown to be on the active chromosome: *** (100%) and ** (approximately 75%) which are either unmethylated (AR and PGK-1) or methylated (DXS255), nt = not tested, r = random. In individual II.5, there is a de novo mutation at the DXS255 locus (see text for further discussion).
Family 2 is a smaller pedigree, which also has the disease present in three generations. The phenotype is mild with clinical signs confined to skin and teeth in I.2 and II.1. Individual III. 1 has left optic atrophy and has no vision in that eye. They are all of normal intelligence. DNA was extracted from blood lymphocytes by standard methods. For the analysis of RFLP markers, DNA was digested with appropriate restriction enzymes according to suppliers’ instructions and fragments separated by agarose gel electrophoresis and transferred to nylon membranes (Hybond-N or Gene-Screen Plus). Probes were labelled with [32P] dTTP by random priming and hybridization carried out in 5 × SSC at 65 °C in the presence of dextran sulphate and Denhart’s solution [13].
PCR-based polymorphisms were analysed in standard reactions of 25 µl containing 250 ng DNA, 0.2 mM dNTPs, each primer at 0.1 µM and 1 unit of Taq polymerase with the appropriate manufacturer’s buffer. 30–34 cycles were carried out to provide enough product for ethidium bromide visualization of alleles. Microsatellite polymorphisms were separated by vertical electrophoresis in native Polyacrylamide gels of 5–10% along with pBR322 DNA digested with HaeIII and MspI as size markers.
Lod scores were calculated using the LINKAGE programme [14] on the basis of a frequency for IP of 0.0001. The allele frequency at the other loci was determined solely by the number of alleles present. The disorder was assumed to be fully penetrant and females showing no clinical signs from birth were scored as unaffected.
Analysis of X chromosome activity was carried out at the AR and PGK-1 genes and at DXS255. At the sites studied in the AR and PGK-1 genes, there is evidence that the active X chromosome is unmethylated while the inactive X chromosome is methylated [15, 16].
The AR locus was examined using primers for a region including the highly polymorphic poly(CAG) repeat in the coding region [17, 18] and a differentially methylated HpaII site using a modification of a published method [15]. 1.4 µg DNA was digested overnight with 10 units of HpaII in the manufacturers’ buffer in a total volume of 30 µl. The enzyme was inactivated by boiling for 5 min and 8 µl used as template for PCR. Control samples of an equivalent amount of DNA which had not been digested with HpaII were also amplified in the standard buffer. 35 pmol each primer [15] was used in a total volume of 50 µl with Mg2+ only added to the control samples. After an initial denaturation for 3 min, amplification was carried out for 34 cycles of 94°C for 1 min, 52°C for 1 min, 72°C for 1 min (extended by 3 s/cycle) with a final extension at 72°C for 10 min. The alleles were separated by electrophoresis on 5% native Polyacrylamide gels stained with ethidium bromide.
At the PGK1 locus, PCR amplification of a region including exon 1, differentially methylated HpaII sites and the BstXI polymorphic site [16, 19] was carried out using the primers A1 and B3 as published [19]. A 10 µl aliquot of amplified DNA was subsequently digested in a 20 µl reaction with 10 units of BstXI and manufacturer’s recommended buffer. The alleles were separated by electrophoresis in a 4% agarose gel (3 parts NuSieve:l part normal) and stained with ethidium bromide.
DXS255 was studied using clone M27ß which detects sites which are methylated on the active and undermethylated on the inactive chromosome [20–22]. Clone M27β was used as a labelled probe against Southern blots of genomic DNA digested with PstI and in double digests with PstI and either MspI or HpaII [23].
Results
The pedigrees of the two families studied are shown in figure 1. Two point linkage data between the IP locus and informative markers in family 1 is presented in table 1. The lod scores suggest linkage to the Xq28 markers DXS52 and F8C (lod = 2.58 and 2.53, respectively at θ = 0). In family 2 (data not shown), F8C contributes a lod score of 0.60 making the combined total for the two families 3.13 (θ = 0). At all other loci studied, including DXS426 in Xpl 1.23, the results in both families indicate significant exclusions (table 1) (data not shown).
Genotypes and assumed X chromosome activity at the three loci studied are given in figure 1. Results of the X chromosome activity study in family 1 using the AR polymorphism are shown in figure 2. The paternal allele, 2, is lost after HpaII digest in all the affected girls indicating that it is unmethylated. It has been demonstrated, at this locus, that the active X chromosome is unmethylated [15]. Therefore it is the maternal chromosome which is inactive in their blood cells. In the unaffected girls two patterns are seen: in II.1 and II.6 the maternal allele, 1, is preferentially digested with HpaII while in II.3 both maternal and paternal alleles are digested to the same extent. It is not known which allele is maternal in origin and which paternal in I.2 and I.3 who are both heterozygous. However, both females show loss of the same allele after HpaII digest.

PCR of the AR trinucleotide repeat polymorphism in control (C) and HpaII digested (H) DNA from females in family 1. The individuals are numbered as in figure 1.
In family 1, only I.2 and 2 of her daughters, II.1 (unaffected) and II.2 (affected), were heterozygous for the PGK-1 polymorphism and could be tested for X chromosome activity using that system (data not shown). In the unaffected girl II.1 both alleles were cut by HpaII indicating that there is a random pattern of X chromosome activity. In individual I.2 and her affected daughter II.2, however, one allele is resistant to Hpall, indicating that in the overwhelming majority of cells the same X chromosome is inactive. In II.2, this allele can be identified as maternal in origin.
Study of X chromosome activity using M27β was possible in the heterozygous females I.2, II. 1, II.2, I.3, II.6 and II.7 (data not shown). Shortage of DNA in individual III. 1 did not allow the X chromosome activity status to be determined at this locus. In individual II.5, a de novo mutation has occurred to give an allele, 2, not present in either mother or father. Correct paternity has been demonstrated for this individual using a (CAC)5 fingerprint probe [24, 25] (data not shown). It was found that the chromosome bearing the new mutant allele was uncut by HpaII, the parental origin of this allele could not be determined. In all the females tested, M27β detected a skewed methylation pattern and, hence, provided evidence of nonrandom X chromosome activity. In II. 1 (unaffected) the maternal X is active in the majority of cells while in II.2 (affected) the parental X is active. Unfortunately, due to the absence of paternal DNA, the source of the alleles cannot be identified in II.6 and II.7 although they show different alleles on the predominantly active chromosome. Both mothers (I.2 and I.3) show nonrandom X chromosome activity with the same chromosome active. The single allele in the males, I.1, II.8 and II.9, was uncut by HpaII as expected.
X chromosome activity in family 2 was studied using only the AR system (fig. 1). All 3 affected females show complete loss after HpaII digest of one allele. In individual I.2 the parental origin of the active X chromosome cannot be determined. For II.1 and III. 1 however, the allele lost after HpaII digestion can be identified as the paternal one indicating that in both individuals the maternally inherited chromosome is inactive in the overwhelming majority of cells.
Discussion
Family 1 shows linkage to markers DXS52 and F8C at Xq28, with lod scores of 2.58 and 2.53 respectively, at θ = 0 (table 1). As it is well established that, in the case of two X-linked loci, a lod score of >2 can indicate linkage [26], this suggests that the defective locus is IP2 [8] in this family. Family 2 also gives a small positive lod score at F8C (0.60, θ = 0) which suggests that the same locus is responsible. This conclusion is supported by the exclusions in both families at all other loci tested (table 1) (data not shown).
In family 1, the results from all three loci tested for X chromosome activity show that individuals I.2 and I.3 have the same active X chromosome (fig. 1), although its parental source could not be determined. In all five affected descendant females in this family the inactive X chromosome has been inherited from their mother, as shown most clearly from the results at the AR locus and supported where analysis was possible at PGK-1 and DXS255. The unaffected females, II.1 and II.6, show skewed X chromosome activity but in both cases it is the predominantly active chromosome which is maternal in origin. A random X chromosome activity pattern is seen in the third unaffected daughter (II.3). On the basis of this information, in conjunction with the linkage data which shows that the unaffected females, II.1, II.3 and II.6, have inherited a different maternal allele at the DXS52 and F8C loci from their affected sisters (data not shown), it can be confirmed that these 3 females are highly unlikely to be IP gene carriers.
In other studies, it has been shown that a significant proportion of normal females have skewed patterns of X chromosome activity [e.g. 27] as was observed in II.1 and II.6. The significance of the results in family 1 is the reproducibility with which it is the maternal chromosome which is the inactive one in the affected daughters.
Results in family 2 are similar to those in family 1. Individuals I.2, II. 1 and III.1 have one, predominantly active chromosome and thus show very pronounced, skewed X chromosome activity. In the case of the latter 2 females the inactive chromosome can be identified as having been inherited from their respective mothers.
Two recent studies provide information about skewed X chromosome activity in families and isolated individuals with IP. Firstly, a study of a family where the mother had IP and the father had haemophilia, due to a defective factor VIII gene [28]. The linkage results suggested that the IP defect was in Xq28 at the IP2 locus, although the lod score was too small to be definitive. The affected individual in this family showed skewed X chromosome activity, with her maternal X being the inactive one. Interestingly (and unfortunately), the activity of her paternal X in the majority of cells was confirmed by the fact that she suffered from haemophilia. Her 2 sisters, who had not inherited IP from their mother, as expected did not manifest haemophilia [28].
Secondly, a study of 31 females with IP [27] showed an increased frequency of skewed X chromosome activity compared to controls with both familial and sporadic cases showing the effect. However, mother/daughter pairs showed no familial consistency of skewed activity. In the one family where both mother and daughter were affected by IP and showed skewed activity, the IP mutation was on the active X. However, there was no genetic information to indicate whether the familial cases were due to a defect at IP2 and possible genetic heterogeneity must be considered when results are pooled from a number of families and individuals. Comparing these results to those in our study, despite its limited sample size, we suggest that there is clear evidence that affected females with IP2 show nonrandom X chromosome activity. At other IP loci, as yet, there is no evidence for such skewed X chromosome activity and, indeed, from the results presented by Harris et al. [27] there may be at least one locus for which this does not occur. When the genes for IP2 and other IP loci are cloned, it will be very interesting to investigate the reasons for the presence or absence of skewed X chromosome activity in the different disorders.
Nonrandom X chromosome activity detected in both our families may be the result of initial nonrandom X inactivation in the PBL precursor cells or the result of selection in these cell lineages. Similar results have been observed in DNA extracted from other tissues and from blood cells in IP patients [12] and in several other X-lmkage disorders, for example Wiskott-Aldrich syndrome [29]. The results with PBL DNA suggest that the IP gene may be normally expressed in blood cells or their precursor as well as in skin fibroblasts.
References
Landy SJ, Donnai D: Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet 1993;30:53–59
Carney RG: Incontinentia pigmenti. A world statistical analysis. Arch Dermatol 1976;112:535–542
Garcia-Dorado J, de Unamuno P, Fernandez-Lopez E, Salazar Veloz J, Armijo M: Incontinentia pigmenti: XXY male patient with a family history. Clin Genet 1990;38:128–138
Gorski JL, Burright EN, Harnden CE, Stein CK, Glover TW, Reyner EL: Localisation of DNA sequences to a region within Xpl 1.21 between incontinentia pigmenti (IP1) X-chromosomal breakpoints. Am J Hum Genet 1991;48:53–64
Harris A, Lakester S, Haan E: The gene for incontinenti pigmentia: Failure of linkage studies using DNA probes to confirm cytogenetic localisation. Clin Genet 1988;34: 16.
Sefiani A, Sinnett D, Abel L, et al: Linkage studies do not confirm the cytogenetic localisation of incontinenti pigmentia on Xp11. Hum Genet 1988;80:282–286
Sinnett D, Sefiani A, Lavergne L, et al: Linkage exclusion of incontinenti pigmentia locus from the short arm of X chromosome by RFLP markers. Genome 1988;30(suppl 1):233.
Sefiani A, Abel L, Heuertz S, et al: The gene for incontinenti pigmentia is assigned to Xq28. Genomics 1989;4:427–429
Sefiani A, M’rad R, Simard L, et al: Linkage relationship between incontinenti pigmentia (IP2) and nine terminal X long arm markers. Hum Genet 1991;86:297–299
Hyden-Granskog C, Salonen R, von Koskull H: Linkage studies in families with hereditary incontinenti pigmentia. Hum Genet 1993, 91:185–189.
Wieacker P, Zimmer J, Ropers HH: X inactivation patterns in two syndromes with probable X-linked dominant, male lethal inheritance. Clin Genet 1985;28:238–242
Migeon BR, Axelman J, de Beuer SJ, Valle D, Mitchell GA, Rosenbaum KN: Selection against lethal alleles in females heterozygous for incontinenti pigmentia. Am J Hum Genet 1989;44:100–106
Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. New York, Cold Spring Harbor Laboratory Press, 1989.
Lathrop GM, Lalouel J: Efficient computations in multilocus linkage analysis. Am J Hum Genet 1984;36:460–465
Allen RC, Zoghbi H, Moseley AM, Rossenblatt HM, Belmont JW: Methylationof Hpall and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X-chromosome inactivation. Am J Hum Genet 1992;51:1229–1239
Gilliland DG, Blanchard KL, Levy J, Perrin S, Bunn HF: Clonality in myeloproliferative disorders: Analysis by means of the polymerase chain reaction. Proc Natl Acad Sci USA 1991;88:6848–6852
Tilley WD, Marcelli M, Wilson JD, McPhaul MJ: Characterisation and expression of a cDNA encoding the human androgen receptor. Proc Natl Acad Sci USA 1989;86:327–331
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH: Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991;352:77–79
Van Kamp H, Jansen R, Willemze R, Fibbe WE, Landegent JE: Studies on clonality by PCR analysis of the PGK-1 gene. Nucleic Acids Res 1991;19:2794.
Boyd Y, Fraser NJ: Methylation patterns at the hypervariable X chromosome locus DXS255 (M27B): Correlation with X-inactivation status. Genomics 1990;7:182–187
Brown RM, Fraser NJ, Brown GK: Differential methylation of the hypervariable locus DXS255 on active and inactive X chromosomes correlates with the expression of a human X-linked gene. Genomics 1990;7:215–221
Hendriks RW, Kraakman MEM, Mensink RGJ, Schuurman RKB: Differential methylation at the 5′ and 3′ CCGG sites flanking the X chromosomal hypervariable DXS255 locus. Hum Genet 1991;88:105–111
Abrahamson G, Fraser NJ, Boyd Y, Craig I, Wainscoat JS: A highly informative X chromosome probe, M27B, can be used for the determination of tumour clonality. Br J Haematol 1990;74:371–372
Schafer R, Zischler H, Epplen JT: (CAC)5, a very informative oligonucleotide probe for DNA fingerprinting. Nucleic Acids Res 1988,16:51–69.
Nurnberg P, Roewer L, Neitzel H, Sperling K, Popperl A, Hundreiser J, Poche H, Epplen C, Zischler H, Epplen JT: DNA fingerprinting with the oligonucleotide probe (CAC)5/(GTG)5: Somatic stability and germline mutations. Hum Genet 1989;84:75–78
Ott J: Analysis of Human Genetic Linkage, rev ed. Baltimore, Johns Hopkins Press, 1991.
Harris A, Collins J, Vetrie D, Cole C, Bobrow M: X inactivation as a mechanism of selection against lethal alleles: Further investigation of incontinenti pigmentia and X-linked lymphoproliferative disease. J Med Genet 1992;29:608–614
Coleman R, Genet SA, Harper JI, Wilkie AOM: Interaction of incontinenti pigmentia and factor VIII mutations in a female with biased X inactivation, resulting in haemophilia. J Med Genet 1993;30:497–500
Hendriks RW, De Weers M, Mensink RG, et al: Diagnosis of Wiskott-Aldrich syndrome by analysis of the X chromosome inactivation patterns in maternal leukocyte populations using the hypervariable DXS255 locus. Clin Exp Immunol 1991;84:219–222
Acknowledgements
We would like to acknowledge the assistance and helpful discussions from all staff of the Molecular Genetics Unit. In particular, we would like to thank Judith Goodship for discussions regarding the study of X chromosome activity and R.C. Allen for providing a preprint of his paper. This research was funded by the Sir Jules Thorn Trust and the Wellcome Trust (Grant No. 033229/Z/91/1/1.27/DG).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Curtis, A.R.J., Lindsay, S., Boye, E. et al. A Study of X Chromosome Activity in Two Incontinentia pigmenti Families with Probable Linkage to Xq28. Eur J Hum Genet 2, 51–58 (1994). https://doi.org/10.1159/000472341
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1159/000472341
