
Abstract
Phenylketonuria (PKU) is an autosomal recessive disorder associated with a deficiency of hepatic phenylalanine hydroxylase (PAH). Although the molecular lesions present in the PAH genes of PKU patients have previously been determined in several Slavic populations, little is known regarding the molecular basis of PKU in the non-Slavic populations of the former Soviet Union. Guthrie card samples from twenty-one classical PKU patients residing in the Tatarian Republic were examined by a combination of denaturing gradient gel electrophoresis and direct sequence analysis. Twelve patients were of Tatarian ancestry, five were of Russian ancestry, and four were of mixed Tatarian and Russian ancestry. Two of the Tatarian patients were related, sharing one mutant allele. The single major allele identified in this study was R408W/RFLP haplotype 2/VNTR 3, which was present on 11/14 or 78.6% of all mutant chromosomes of Slavic origin, but on only 10/27 or 37.0% of mutant chromosomes of Tatarian origin. This result suggests that this allele was introduced into central Asian populations during the eastward expansion of Slavs across the Eurasian landmass. A significant influence of Turkic peoples on Tatars can be inferred from the presence of R261Q, IVS10nt546g→a, L48S, IVS2nt5g→c: and P281L, all of which are relatively common among Turks or have been observed in Mediterranean populations. Together, these alleles are present on 11/27 or 40.7% of all mutant chromosomes in ethnic Tatars. Surprisingly, the common Scandinavian mutation IVS12nt 1g→a was also present in Tataria, as was the ΔagE221D222fs mutation found previously only in Denmark. Thus, some direct or indirect gene flow from Scandinavian into Tataria seems evident. Finally, the complete absence of PAH mutations previously observed in Oriental populations suggests that there was little gene flow into Tataria from Eastern Asia.
Similar content being viewed by others


Introduction
Phenylketonuria (PKU) is an autosomal recessive disorder in which individuals are unable to hydroxylate the essential amino acid L-phenylalanine. Severe mental retardation develops unless phenylalanine intake is restricted [1]. Most individuals with PKU carry mutations in the phenylalanine hydroxylase (PAH) locus, where over 200 mutations have been identified so far [2–5]. Most of these mutations are strongly associated with specific RFLP [2, 3] or VNTR [6] haplotypes. The R408W/RFLP haplotype 2/VNTR haplotype 3 (R408W/HT 2/VNTR 3) allele has been identified as the major cause of PKU in Slavic populations (i.e. those modern human populations derived from Slavic-speaking peoples) [7–9], and both Slavic [8–10] and ‘Balto-Slavic’ [7] origins have been proposed. An alternative hypothesis is that this mutant allele arose among various ethnic populations of central or east Asia, and was subsequently introduced into Slavic or Baltic peoples. An origin for R408W/HT 2/VNTR 3 among the Mongoloid peoples of east Asia can be discounted, since this allele has not been detected among Chinese or Japanese PKU patients [Eisensmith, unpubl. obs.]. However, an origin among Tatars or other indigenous central Asian peoples cannot be completely dismissed in the absence of data concerning the relative frequency of the R408W/HT 2/VNTR 3 allele in these populations.
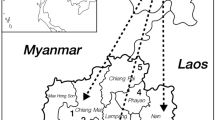
This possibility was examined by determining the relative frequencies and haplotype associations of R408W and other PAH mutations among PKU patients residing in the Tatarian Republic of the Russian Federation, a region located primarily south and east of the city of Kazan (fig. 1). The population in this region is nearly evenly divided between ethnic Russians and Tatarians (44 vs. 48%, respectively) [11], and the incidence of PKU is approximately 1:6,000 in both populations. The relative frequency of the R408W/HT 2/VNTR 3 allele was significantly greater among mutant chromosomes of Slavic origin than those of Tatarian origin, further corroborating previous studies suggesting a Slavic origin for this allele [7–10]. Other PAH mutations common among mutant chromosomes of Tatarian origin were those previously observed in Turks or other Mediterranean populations, suggesting a strong genetic influence of Turkic peoples on modern Tatars. No PAH mutations previously observed in east Asian populations were detected in this study, suggesting little or no gene flow from this region into Tataria.

Human migration into Tataria. The hatched area indicates the historical region of Tatarian settlement from the tenth through the twentieth centuries. The solid arrows indicate the various waves of human migration into this region. The sizes of the different arrows represent estimates of the magnitude of the influx into Tataria from the different sources.
Materials and Methods
Patients
From newborn screening and retrospective analysis of serum phenylalanine levels in mentally retarded patients, the incidence of PKU among both ethnic Russians and Tatarians residing in the Tatarian Republic is approximately 1 in 6,000 [Sergeeva, unpubl. obs.]. Using 1980 census data regarding the size and ethnic composition of the Tatarian Autonomous Republic [11], this incidence would yield a total of approximately 250 PKU patients of Russian and Tatarian ancestry. However, after termination of treatment at age 6, most of these affected individuals do not return to the clinic and are therefore not available for mutation studies. Guthrie cards from 21 individuals with classical PKU were collected at the Medical Genetics Center in Kazan during a 1-month period in 1992. Classical PKU was defined in these patients by the presence of severe mental retardation and serum phenylalanine levels greater than 1,200 µM in untreated individuals, or by similarly elevated serum phenylalanine levels during the neonatal period prior to treatment. All patients or their parents were informed of the nature of the study. Consent was obtained for collection of blood samples for mutation analysis and for review of the medical records. All procedures were performed in accordance with the guidelines and approval of the Institutional Review Boards of both medical centers.
Twelve patients were of Tatarian ancestry, five were of Russian ancestry, and four were of mixed Tatarian and Russian ancestry. Two Tatarian patients were related, sharing one mutant allele. Chromosomes were defined as Tatarian in origin if they were transmitted from individuals who had Tatarian surnames, who spoke the Tatarian language, or who claimed at least three generations of Tatarian ancestors during interview. Similar criteria were used to identify Russian chromosomes.
Primer Design
Due to the relatively low yield of PCR products obtained from some Guthrie cards, it was often necessary to perform a two-step amplification procedure using three different oligonucleotide primers. Complete primer sequences for all thirteen exons are listed in table 1. All primers were designed using the OLIGO-Primer Analysis Program [12] to minimize cross homology and self-complementation. The GC-clamp sequence was identical to that reported by Guldberg et al. [13]. Each exon of gene was first amplified using an appropriate pair of flanking primers (primers A and B in table 1). A second amplification reaction was then performed using either primer A or B from the first reaction and a third primer (primer C in table 1) located internal to A and B that contained a GC clamp. This two-step procedure resulted in a slight rise in the background signal due to the presence of Taq-generated mutations, but the specific bands associated with the normal and mutant alleles and their heteroduplexes could always be easily distinguished from the background.
PCR Amplification Protocol
The PCR reaction mixture (0.2-mm2 piece of the Guthrie card containing the dried whole blood, 200 µM dNTPs, 1.7 mM Mg2+, and 0.3 µM each of primers A and B in a volume of 10 µl) was first denatured for 15 min at 97 °C in a Perkin-Elmer Model 9600 Thermocycler. 0.2 U of Taq DNA polymerase in a volume of 10 µl were added at 85 °C, and the PCR reaction proceeded for 20 cycles consisting of 1 min at 55°C, 1 min at 72°C and 10 s at 94°C. 0.3 U of Taq DNA polymerase and 15 µM each of the GC-clamped primer C and its opposite primer (A or B, depending on the exon) in a volume of 30 µl were then added to the reaction tubes at 85°C, and the PCR reaction proceeded for another 40 cycles consisting of 10 s at 55°C, 20 s at 72°Cand 10 s at 94°C.
Denaturing Gradient Gel Electrophoresis (DGGE) Protocol
Ten to twenty microliters of the GC-clamped PCR product were loaded onto a 6% Polyacrylamide gel containing a gradient of urea and formamide ranging from 20 to 80% [13]. Electrophoresis was performed at 150 V for 6 h in a 61 °C TAE solution (0.04 M Trisacetate, 1 mM EDTA, pH 8.0) contained in a DGGE apparatus obtained from CBS Scientific (Del Mar, Calif., USA).
Sequencing Protocol
PCR products were first purified from agarose gel using the QIAquick gel extraction kit (Qiagene, Hilden, Germany). Double-stranded DNA sequencing was performed using the Taq DyeDeoxy Terminator Sequencing kit and sequencing gels were analyzed on a 373A automated sequencing machine (Applied Biosystems, Foster City, Calif., USA). In some cases, the abnormal band corresponding to the mutant homoduplex was first cut from the Polyacrylamide gel after DGGE and reamplified using primers A or B and C as described above. The amplified product was then extracted and sequenced as described previously.
VNTR and RFLP Haplotype Analysis
VNTR haplotypes were determined as previously described [6]. The RFLP sites that could be examined by PCR-based methods were analyzed as previously described (BglII. [14]; PvuIIa [15]; PvwIIb [Eisensmith, to the PAH Gene Mutation Analysis Consortium, 1993]; MspI [16]; XmnI [17]).
Results
Twenty-one Tatarian PKU patients representing forty-one independent alleles were examined by a combination of DGGE and direct sequence analysis. Twelve different alleles containing 10 different mutations were identified, and their relative frequencies among ethnic Russians, Tatars and the total sample population are listed in table 2. The major mutation observed in both the Russian (11/14, 78.6%) and Tatarian (10/27, 37.0%) populations was R408W. The significantly higher frequency observed in Russians versus Tatars provides additional evidence for a Slavic or Baltic origin for R408W [7–9].
The twenty non-R408W alleles in these populations could be roughly divided into three classes: alleles containing mutations previously observed in Turks or other peoples of Mediterranean ancestry, including R261Q [18–19], L48S [20], IVS2nt5g→c [21], IVS10nt546g→a [22–24] and P281L [25, 26]; alleles containing mutations of known or presumed Scandinavian origin, such as the IVS12nt 1g→a [27] and ΔagE221D222fs [28] mutations, and alleles containing the moderately prevalent European mutation R158Q [19,29].
In addition to these ten mutations, six polymorphisms were also detected by DGGE analysis. Two of these, S137S and IVS12nt-35g→a, were novel, while IVS2nt19t→c [30], Q232Q [31], V245V [32] and L385L [31] have now been observed in many different populations. The associations between these polymorphic markers in the PAH gene and the PAH mutations observed in this study are shown in table 3. Most of the associations observed were absolutely inclusive and compatible with known VNTR or RFLP haplotype associations in other populations (data not shown). For example, both IVS10nt546 mutations were associated with the common polymorphism IVS2nt19 and with the 7-copy VNTR allele (table 3). A further examination of the RFLP sites that could be amplified by PCR-based methods (data not shown) narrowed the haplotype association for this mutation to 6 or 36 and excluded an association with 10, 34, or other haplotypes known to be associated with IVS10nt546 [5].
In contrast, on the basis of PCR analyses (data not shown), R158Q and R261Q were each associated with two different RFLP/VNTR haplotypes. One chromosome containing the R158Q mutation and the 8-copy VNTR allele (R158Q/VNTR 8 in tables 2 and 3) was apparently associated with an RFLP haplotype other than those previously reported for this mutation, namely haplotypes 4 [19, 29], 16 and 28 [33], while the available data for the second chromosome containing R158Q and the 3-copy VNTR allele (R158Q/VNTR 3 in tables 2 and 3) are compatible with RFLP haplotypes 4 or 28. Of the four chromosomes bearing the R261Q mutation, three also contained seven VNTR repeats. This mutation/VNTR association (R261Q/VNTR 7 in tables 2 and 3) has previously been observed on only one chromosome from Germany [6], where R261Q was present on a haplotype 1 background. The fourth chromosome bearing R261Q contained three copies of the VNTR (R261Q/VNTR 3 in tables 2 and 3), suggesting an association with RFLP haplotype 2, as previously described among a southwest-European population by Caillaud et al. [34]. At present, there are too little data and too few alleles to determine whether these different haplotype associations are the result of recurrent mutation or other mechanisms such as crossover or gene conversion.
Discusssion
A combination of DGGE and direct sequence analysis was used to identify the PAH mutations present among twenty-one classical PKU patinets residing in the Tatarian Republic of the Russian Federation. Although only dried blood spots were available as a source of genomic DNA, modifications of previously published methods [13, 28] permitted the determination of 100% of the mutations present in this ethnically mixed patient population. Twelve different mutant alleles carrying ten different mutations were found, as were six different silent polymorphisms. A comparison of the distribution of mutations and polymoronisms at the PAH locus revealed significant differences between ethnic Russians and Tatars. As observed in previous studies of Slavic populations of eastern Europe [7–10], the predominant allele among Russian PKU patients was R408W/HT 2/VNTR 3. In the present study, this allele accounted for nearly 80% (11/14) of all mutant alleles in the small, ethnic Russian subpopulation of Tataria. The second most frequent allele among ethnic Russians (2/14 or 14.3%) was the common northern-European allele IVS12nt1. The presence of this allele among ethnic Russians residing in Tataria supports the previous findings of Charikova et al. [35], who reported a frequency of approximately 16% for this mutation among PKU Patients collected from the Moscow region. The final mutation observed among ethnic Russians was R270K; the only available data regarding this mutation is a report of its presence on a chromosome of a Portuguese patient [Leandro, to the PAH Gene Mutation Analysis Consortium, 1993] and on one chromosome of an American PKU patient of undetermined ancestry [Guldberg, to the PAH Gene Mutation Analysis Consortium, 1993].
As in other populations [8, 9, 36–39], the relative frequency of PAH mutations in Tataria may provide interesting and useful corroborating evidence for human migration based on previous genealogical, historical or archaeological studies. The history of Tataria extends from the tenth through the twentieth centuries and incorporates the migrations of several different peoples into this region. Different waves of migration brought Bulgars, Slavs and Swedes into the middle Volga region from the west or south. Later migrations into Tataria brought Kypchak Turkic tribes from the Altai region of central Asia (fig. 1) [40]. The genetic contributions of these different migrations may be reflected in the spectrum of PAH mutations present in modern Tatars. For example, the relatively high frequency of the common Slavic PKU allele R408W/HT 2/VNTR 3 in Tatars is consistent with the historical evidence of a lengthy period (> 1,000 years) of Slavic influence in Tataria [41]. Similarly, the high combined frequency of PAH mutations previously observed in Turks or other peoples of Mediterranean ancestry (R261Q [18, 19], L48S [20], IVS2nt5g→c [21], IVS10nt546g→a [22–24] and P281L [25, 26]) corroborates historical and linguistic evidence [40] for significant contributions of Turkic peoples to the gene pool of modern Tatars. Whether these mutations were transmitted from Bulgars, Kypchak Turks or some combination of these two groups remains to be determined. The complete lack of PAH mutations previously observed among the Chinese, Japanese, Koreans or Taiwanese indicates that little or no gene flow occurred into Tataria from the the northern Mongoloid peoples of east Asia.
Somewhat surprising in light of the estimated sizes of previous migrations into Tataria is the presence of mutations of putative Scandinavian origin. These include the major Scandinavian mutation IVS12nt1 [8] and ΔagE221D222fs. Together, these mutations accounted for about 15% of all PKU alleles in Tatars. Two different hypotheses could account for the presence of these mutations in Tatars. One possibility is indirect gene flow from Scandinavians to Tatars. According to this hypothesis, the IVS12ntl mutation could have been transmitted into Tatars through Slavic ancestors. The common ΔagE221D222fs mutation could also have been transmitted from Scandinavians via this indirect route, or could have been introduced into Tatars from another ethnic group. It is difficult to establish the origin of this mutation from only a single previous occurrence [28]. A second possibility is that both of these mutations are in fact Scandinavian in origin and were introduced directly into Tatars. Archaeological and historical evidence indicates that Norse tribes migrated south and east across the eastern European landmass beginning in the 4th century BC [42]. Later records indicate that some groups of Swedish Vikings who utilized the Volga as a major trade route to Persia settled in Tataria during the latter part of the first millennium [43]. No evidence other than the present study exists to suggest whether the size of these migrations was sufficient to leave genetic traces, and resolving these indirect or direct hypotheses will require additional data from other genetic loci.
References
Scriver CR, Kaufman S, Eisensmith RC, Woo SLC: The hyperphenylalaninemias; in Scriver CR, Beaudet AL, Sly WS, Valle D (eds): The Metabolic and Molecular Bases of Inherited Disease, ed 7. New York, McGraw-Hill, 1995, vol 1, pp 1015–1075.
Eisensmith RC, Woo SLC: Molecular basis of phenylketonuria and related hyperphenylalaninemias: Mutations and polymorphisms in the human phenylalanine hydroxylase gene. Hum Mut 1992;1:13–23.
Scriver CR, John SWM, Rozen R, Eisensmith R, Woo SLC: Associations between populations, PKU mutations and RFLP haplotypes at the PAH locus: An overview. Dev Brain Dysfunction 1993;6:11–25.
Eisensmith RC, Woo SLC: Molecular genetics of phenylketonuria: From molecular anthropology to gene therapy. Adv Genet 1995;32: 199–271.
Scriver CR (ed): PAH Gene Mutation Analysis Consortium. April 1994. McGill University, Montreal.
Goltsov AA, Eisensmith RC, Konecki DS, Lichter-Konecki U, Woo SLC: Linkage disequilibrium between mutations and a VNTR in the human phenylalanine hydroxylase gene. Am J Hum Genet 1992;51: 627–636.
Kalaydjieva L, Dworniczak B, Kucinskas V, Yurgeliavicius V, Kunert E, Horst J: Geographical distribution gradients of the major PKU mutations and the linked haplotypes. Hum Genet 1991;86:411–413.
Eisensmith RC, Okano Y, Dasovich M, Wang T, Güttler F, Lichter-Konecki U, Konecki DS, Svensson E, Hagenfeldt L, Rey F, Munnich A, Lyonnet S, Cockburn F, Conner JM, Pembrey ME, Smith I, Gitzelmann R, Steinmann B, Apold J, Eiken HG, Giovannini M, Riva E, Longhi R, Romano C, Cerone R, Naughten ER, Mullins C, Cahalane S, Özalp I, Fekete G, Schuler D, Berensci GY, Nász I, Brdicka R, Kamaryt J, Pijackova A, Cabalska B, Bozkowa K, Schwartz E, Kalinin VN, Jin L, Chakraborty R, Woo SLC: Multiple origins for phenylketonuria in Europe. Am J Hum Genet 1992;51:1355–1365.
Eisensmith RC, Goltsov AA, O’Neill C, Tyfield LA, Schwartz EI, Kuzmin AI, Baranovskaya SS, Tsukerman GL, Treacy E, Scriver CR, Güttier F, Guldberg P, Eiken HG, Apold J, Svensson E, Naughten E, Cahalane SF, Croke DT, Cockburn F, Woo SLC; Recurrence of the R408W mutation in the PAH locus in Europeans. Am J Hum Genet 1995;56:278–286.
Jaruzelska J, Matuszak R, Lyonnet S, Rey F, Rey J, Filipowicz J, Borski K, Munnich A: Genetic background of clinical heterogeneity of phenylketonuria in Poland. J Med Genet 1993;30:232–234.
Simon G: Nationalism and Policy Toward the Nationalities in the Soviet Union: From Totalitarian Dictatorship to Post-Stalinist Society. Boulder, Westview, 1991, p 376.
Rychlik W, Rhoads RE: A computer program for choosing optimal oligonucleotide primers for filter hybridization, sequencing and in vitro amplification. Nucleic Acids Res 1989; 17:8543–8551.
Guldberg P, Romano V, Ceratto N, Bosco P, Ciuna M, Indelicate A, Mollica F, Meli C, Giovannini M, Riva E, Biasucci G, Henriksen KF, Güttier F: Mutational spectrum of phenylalanine hydroxylase deficiency in Sicily: Implications for diagnosis of hyperhphenylalaninemia in southern Europe. Hum Mol Genet 1993;2:1703–1707.
Dworniczak B, Wedemeyer N, Horst J: PCR detection of the BglII RFLP at the human phenylalanine hydroxylase (PAH) locus. Nucleic Acids Res 1991; 19:1958.
Dworniczak B, Wedemeyer N, Eigel A, Horst J: PCR detection of the PvuII (Ea) RFLP at the human phenylalanine hydroxylase (PAH) locus. Nucleic Acids Res 1991; 19: 1958.
Wedemeyer N, Dworniczak B, Horst J: PCR detection of the MspI (Aa) RFLP at the human phenylalanine hydroxylase (PAH) locus. Nucleic Acids Res 1991;19:1959.
Goltsov AA, Eisensmith RC, Woo SLC: Detection of the XmnI RFLP at the human phenylalanine hydroxylase locus by PCR. Nucleic Acids Res 1992;20:927.
Abadie V, Lyonnet S, Maurin S, Berthelon M, Caillaud C, Giraud F, Mattei JF, Rey J, Rey F, Munnich A: CpG dinucleotides are mutation hot spots in phenylketonuria. Genomics 1989;5:936–939.
Okano Y, Wang T, Eisensmith RC, Steinmann B, Gitzelmann R, Woo SLC: Missense mutations associated with RFLP haplotypes 1 and 4 of the human phenylalanine hydroxylase gene. Am J Hum Genet 1990;46:18–25.
Konecki DS, Schlotter M, Trefz FK, Lichter-Konecki U: The identification of two mis-sense mutations at the PAH gene in a Turkish patient with phenylketonuria. Hum Genet 1991;87:389–393.
Kalaydjieva L, Dworniczak B, Kremensky I, Radeva B, Horst J: Population genetics of phenylketonuria in Bulgaria. Dev Brain Dysfunct 1993; 6:39–45.
Dasovich M, Konecki D, Lichter-Konecki U, Eisensmith RC, Guttler F, Naughton E, Mullins C, Woo SLC: Molecular characterization of a PKU allele prevalent in southern Europe and Ireland. Somat Cell Mol Genet 1991;17:303–309.
Dworniczak B, Aulehla-Scholz C, Kalaydijeva L, Ullrich K, Bartholome K, Grudda K, Horst J: Aberrant splicing of phenylalanine hydroxylase mRNA: The major cause for phenylketonuria in parts of southern Europe. Genomics 1991; 11:242–246.
Kalaydjieva L, Dworniczak B, Aulehla-Scholz C, Devoto M, Romeo G, Sturhmann M, Horst J: Phenylketonuria mutation in southern Europeans. Lancet 1991;337:865.
Okano Y, Wang T, Eisensmith RC, Longhi R, Giovannini M, Cerone R, Romano C, Woo SLC: Phenylketonuria missense mutations in the Mediterranean. Genomics 1991;9: 96–103.
Dworniczak B, Grudda K, Stümper J, Bartholomé K, Aulehla-Scholz C, Horst J: Phenylalanine hydroxylase gene: Novel missense mutation in exon 7 causing severe phenylketonuria. Genomics 1991;9:193–199.
DiLella AG, Marvit J, Lidsky AS, Guttler F, Woo SLC: Tight linkage between a splicing mutation and a specific DNA haplotype in phenylketonuria. Nature 1986;322:799–803.
Guldberg P, Henriksen KF, Güttler F: Molecular analysis of phenylketonuria in Denmark: 99% of the mutations detected by denaturing gradient gel electrophoresis. Genomics 1993;17:141–146.
Dworniczak B, Aulehla-Scholz C, Horst J: Phenylketonuria: Detection of a frequent haplotype 4 allele mutation. Hum Genet 1989;84:95–96.
Lichter-Konecki U, Schlotter M, Konecki DS: DNA sequence polymorphisms in exonic and intronic regions of the human phenylalanine hydroxylase gene aid in the identification of alleles. Hum Hered 1994; 94:307–310.
Forrest SM, Dahl HH, Howells DW, Dianzani I, Cotton RGH: Mutation detection in phenylketonuria using chemical cleavage of mismatch: Importance of using probes from both normal and patient samples. Am J Hum Genet 1991;49:175–183.
Dworniczak B, Aulehla-Scholz C, Horst J: Phenylalanine hydroxylase gene: Silent mutation uncovers evolutionary origin of different alleles. Clin Genet 1990;38:270–273.
Abadie V, Lyonnet S, Melle D, Bertheion M, Caillaud C, Labrune P, Rey F, Rey J, Munnich A: Molecular basis of phenylketonuria in France. Dev Brain Dysfunct 1993;6: 120–126.
Caillaud C, Lyonnet S, Melle D, Rey F, Bertheion M, Vilarinho L, Vas Osorio R, Rey J, Munnich A: Molecular heterogeneity of mutant haplotype 2 alleles in phenylketonuria. Am J Hum Genet 1990;47:A152.
Charikova EV, Khalchitskii SE, Antoschechkin AG, Schwartz EI: Distribution of some point mutations in the phenylalanine hydroxylase gene of phenylketonuria patients from the Moscow region. Hum Hered 1993;43:244–249.
Lyonnet S, Melle D, De Braekeleer M, Laframboise R, Rey F, John SWM, Bertheion M, Berthelot J, Journel H, Le Marec B, Parent P, de Parscau L, Saudubray JM, Rozen R, Rey J, Munnich A, Scriver CR: Time and space clusters of the French-Canadian M1V phenylketonuria mutation in France. Am J Hum Genet 1992;51:191–196.
Perez B, Desviat LR, Die M, Cornejo V, Chamoles NA, Nicolini H, Ugarte M: Presence of the Mediterranean PKU mutation IVS10 in Latin America. Hum Mol Genet 1993; 2:1289–1290.
Treacy E, Byck S, Clow C, Scriver CR: ‘Celtic’ phenylketonuria chromosomes found? Evidence in two regions of Quebec province. Eur J Hum Genet 1993;22:220–228.
Rozen R, Mascisch A, Lambert M, Laframboise R, Scriver CR: Mutation profiles of phenylketonuria (PKU) in Quebec populations: Evidence of stratification and novel mutations. Am J Hum Genet 1994; 55:321–326.
Menges KH: The Turkic Languages and Peoples: An Introduction to Turkic Studies. Wiesbaden, Harrassowitz, 1968.
Rorlich AA: The Volga Tatars. Stanford, Hoover Institution Press, 1986.
Vasiliev AA: The Goths in the Crimea. Cambridge, Mediaeval Academy of America. 1936.
Vernadsky G: Ancient Russia. New Haven, Yale University Press, 1943.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Kuzmin, A.I., Eisensmith, R.C., Goltsov, A.A. et al. Complete Spectrum of PAH Mutations in Tataria: Presence of Slavic, Turkic and Scandinavian Mutations. Eur J Hum Genet 3, 246–255 (1995). https://doi.org/10.1159/000472305
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1159/000472305
