Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain
the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in
Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles
and JavaScript.
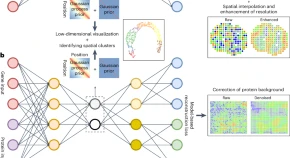
Using a dependency-aware deep generative framework, spaVAE efficiently models spatially resolved transcriptomics data and advances diverse analysis tasks. Following similar strategies, spaPeakVAE and spaMultiVAE enable spatial ATAC-seq data and spatial multi-omics data modeling and analysis, respectively.
The Consortium for Top-Down Proteomics conducted a study to develop and test protocols for native mass spectrometry combined with top-down fragmentation of proteins and protein complexes across eleven instruments in nine laboratories. They report the summary of the outcomes and their recommendations in this Analysis.
OpenFold is a trainable open-source implementation of AlphaFold2. It is fast and memory efficient, and the code and training data are available under a permissive license.
By effective and efficient integration of PacBio HiFi, Oxford Nanopore Technologies ultra-long and other sequencing data types, hifiasm (UL) enables telomere-to-telomere diploid and polyploid genome assembly at a population scale.
By learning to embed DNA k-mers and cells into a joint space, CellSpace improves single-cell ATAC-seq analysis in multiple tasks such as latent structure discovery, transcription factor activity inference and batch effect mitigation.
Single-cell quantitative expression reporters enable high-sensitivity quantitative characterization of cis-regulatory elements at the single-cell level in multicellular systems.
Molecular Pixelation is an optics-free method that uses DNA-tagged antibodies to enable identification of the relative location of proteins on single cells.
Combining genome-wide CRISPR–Cas9-mediated base editors with temporally resolved phosphoproteomics enables the functional screening of thousands of post-translational modification sites involved in T cell activation.
ESI-cryoPrep is a cryo-EM specimen preparation method that employs electrospray ionization techniques to deposit charged macromolecule-containing droplets on EM grids. Demonstrated across various protein samples, this approach effectively prevents biomolecule adsorption at air–water or graphene–water interfaces, addressing challenges related to protein denaturation and preferred orientation.
Super-resolution imaging of reference and target structures enables precise determination of the labeling efficiency of high-affinity binding proteins in cells for improved quantitative assessment of protein organization at the single-molecule level.
Generating training data for training deep-learning-based tools is time consuming. The DELiVR pipeline facilitates this process as demonstrated in this study on detecting c-Fos+ cells or microglia in the brain, following tissue clearing and imaging with light-sheet microscopy.
LiMCA offers a tool for co-profiling 3D genome structure and gene expression at the single-cell level, enabling researchers to elucidate the olfactory receptor gene selection process.
A pretrained foundation model (UniFMIR) enables versatile and generalizable performance across diverse fluorescence microscopy image reconstruction tasks.
brainlife.io is a one-stop cloud platform for data management, visualization and analysis in human neuroscience. It is web-based and provides access to a variety of tools in a reproducible and reliable manner.
The CPJUMP1 Resource comprises Cell Painting images and profiles of 75 million cells treated with hundreds of chemical and genetic perturbations. The dataset enables exploration of their relationships and lays the foundation for the development of advanced methods to match perturbations.
scGHOST offers a computational tool to annotate single-cell subcompartments from scHi-C or imaging data through graph representation learning with constrained random walk sampling.