
Abstract
Birefringence and tensile stress of vulcanized natural rubber samples were measured simultaneously during fast deformation and subsequent holding at a prefixed extension ratio. The deviation from the stress-optical law due to finite chain extensibility could be clearly detected, which became pronounced with an increase in true stress. The increase in birefringence during the holding process was related to the different degrees of molecular-chain orientation to be incorporated into strain-induced crystals according to the network-chain density and the strain ratio of holding. During the holding process, normalized stress decreased linearly with the logarithm of elapsed time in a manner that is almost independent of network-chain density. These experimental results could be successfully explained by assuming a certain fraction of network chains that behave in a manner similar to that of a fluid.
Similar content being viewed by others
Introduction
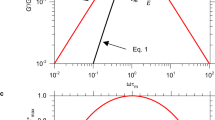
In previous studies on strain-induced crystallization of vulcanized natural rubber (NR) and its synthetic analog (cis-1,4-polyisoprene rubber; IR), the following three peculiar features have been reported. The first feature is the coexistence of highly stretched molecular chains and randomly coiled ones in the extended samples.1, 2, 3, 4, 5, 6, 7, 8 The second feature is with regard to the incipient strain of crystallization during the stretching process, which was almost independent of network-chain density in some studies (Figure 1).6, 7, 8, 9, 10, 11, 12 This feature has been inconsistent with predictions in previous treatments on the basis of the statistical theory of rubber elasticity.13 The third feature is the dependence of stress relaxation on strain-induced crystallization. When rubber samples were extended and kept at a constant length, the rate of strain-induced crystallization changed according to the difference of the material (NR or IR) and to network-chain density. The difference in the rate of crystallization between NR and IR was reflected in the rate of stress relaxation. However, among the samples made of NR or IR, the difference in the rate of crystallization due to network-chain density did not affect the rate of stress relaxation.8, 14, 15 One of the authors of this article focused on these features in his recent review article.8

Development of strain-induced crystallization in (a) NR and (b) IR samples having different network-chain densities. The ordinate indicates the integrated intensity of the 200 reflection (I200) in the X-ray diffraction pattern and the abscissa is the strain ratio (α). The smaller value of the sample code indicates the higher network-chain density. Because ‘crystallinity index’ values have considerably fluctuated in the original article,6 I200 values were newly estimated for this figure from the original data. The details of the experimental procedures and specimens are written in Tosaka et al.6
To explain these features of strain-induced crystallization consistently with earlier theories,13, 16, 17 the primitive mechanical model by James and Guth,18 which assumes elastically effective network chains and fluid-like ones, was reconsidered.19 This ‘two-component model’ could reproduce the experimental features of strain-induced crystallization observed by the wide-angle X-ray diffraction technique successfully. However, it is not clear whether this model is comprehensively applicable to the crystallization behavior observed in other experimental techniques as well.
For example, birefringence measurement is a technique to assess the average orientation of molecular chains. In the classic studies on rubber elasticity, the relationship between birefringence and true stress (namely, force divided by the actual cross-sectional area of the deformed specimen) has been explored. When the tensile strain is small compared with the network-chain length, the molecular network is assumed to be in the Gaussian region. In this case, the linear relationship between birefringence and true stress;

has been analytically derived, which is known as the stress-optical law.13 In Equation (1), Δn is the birefringence, σt is the true stress and C is the stress-optical coefficient that is independent of network-chain density. The value of C has been estimated to be ca. 1.9 × 10−9 Pa−1 for cis-1,4-polyisoprene (namely, NR and IR).13 Deviations from the stress-optical law sometimes occur because of strain-induced crystallization and/or because of the finite extensibility of network chains, as will be discussed later.
In this study, vulcanized NR samples were extended to a prefixed extension ratio, and then tensile force and birefringence were measured simultaneously. The behaviors of strain-induced crystallization and the corresponding stress relaxation are discussed on the basis of Δn−σt diagrams, along with the two-component model.19
Experimental procedure
Preparation of rubber specimens
Sheets of NR samples (ca. 1 mm thick) were prepared in the same way as in our previous publications.6, 7, 14, 15 The recipes and cure conditions along with the network chain densities of the samples are listed in Table 1. It is convenient for the readers to remember that the smaller number in the sample code corresponds to the shorter network-chain length, that is, the larger network-chain density. Ring-shaped specimens were die cut from sheets and used for the experiments. The inner and outer diameters of the specimens were 14.9 and 16.9 mm, respectively (ca. 50 mm circumference).
Tensile measurements
A conventional tensile tester with a load cell of 100 N capacity was used for uniaxial tensile measurements. The tensile tester equipped a custom-made specimen chuck that enabled optical observation of a fixed part of the specimen in a similar way as in Toki et al.20 To minimize the development of strain-induced crystallization during deformation, we adopted the maximum deformation rate of the instrument. Thus, specimens were stretched to a prefixed strain ratio, αs, at 1000 mm min−1 (0.667 s−1), held at the length for 1 h and retracted to the original length. In this article, the strain ratio, α, is defined as l/l0; l and l0 are the lengths of the deformed and initial states of the sample, respectively. Tensile force was recorded every 0.5 s. σt values were estimated on the basis of the calculated cross-sectional area assuming a constant volume and uniform deformation of the specimen. The ambient temperature was controlled at 22±1 °C.
Birefringence measurements
An optical bench for birefringence measurement was situated across the tensile tester to observe specimens while taking tensile measurements. A white halogen lamp was used as the light source. Spectra of a sample passing the crossed nicols were recorded using a spectrometer (BTC112E; B&W Tek, Newark, DE, USA). The stretching direction of the specimen was rotated by ±45° with respect to the polarization axis of the polarizer and the analyzer. In this case, the observed spectrum is formulated as

where λ is the wavelength, I(λ) is the observed spectrum, I0(λ) is the spectrum of the light source, IC(λ) is the transmittance (namely, the inherent color) of the rubber material and T(λ) is the transmittance due to birefringence.
Necessary information for the estimation of retardation is T(λ), which is obtained by normalization of I(λ) using a reference spectrum, I0(λ)·IC(λ).
Depending on the retardation of a specimen, T(λ) has minima when

and maxima when

where Γ is the retardation. When we observe n sequential maxima between two maxima at wavelengths λ1 and λ2 (λ1>λ2), there is a relation,

which leads to

The same equation is obtained if we select two minima instead of maxima. On the basis of Equation (5), we estimated the retardation of the specimen from the wavelengths of two separated maxima (or minima) in T(λ). Finally, we obtained Δn from Γ as

When the change of Δn was small and fluctuated in an unreasonable manner, a different method was adopted. For a spectrum at an appropriate elapsed time, Γ was estimated using Equation (5). The value of m in Equation (3) was then estimated for a suitable peak. Thereafter, the values of Γ were estimated using m and λ for the selected peak in the succeeding spectra.
Sample thickness was calculated from the initial thickness and the strain ratio in the stretching direction, assuming a constant volume and uniform deformation of the specimen.
There is an advantage to this method compared with other methods using monochromatic light.21, 22, 23, 24 Because this method is relatively insensitive to the clarity of samples, estimation of birefringence is possible even when the specimen becomes turbid because of crystallization. Furthermore, the problem of multiple order (namely, the case of m>1) in highly birefringent materials can be eliminated.25 On the other hand, there may be a disadvantage to this method, which is the fluctuation of the estimated values of Δn due to the wavelength dispersion of the refractive index. (By using more number of peak wavelength values, we can approximate the wavelength dependence of Δn with a polynomial equation.25, 26 If we estimate the Δn values at a constant wavelength using the equation, the fluctuation of data could be avoided. In this study, however, such a time-consuming process was not adopted because we had to process hundreds of spectra.)
Results and discussion
Before presenting the experimental results, the interpretation of the Δn−σt diagram is briefly explained using Figure 2. When the molecular network is in the Gaussian region, the stress-optical law is preserved. In this case, Δn changes linearly with σt (along the straight arrow starting from the origin). However, when the extension ratio is relatively high, the stretched network chains cannot behave as the Gaussian ones because of limited chain extensibility. Once the chains adopt a rather stretched conformation, additional increase in Δn is suppressed despite the increase in σt. As a result, the data point in the Δn−σt diagram tends to drift from the linear relationship to the right side in the case of Figure 2. This type of effect due to finite chain extensibility has been reported for polymer melt27, 28, 29, 30 and cross-linked poly(dimethylsiloxane) filled with silica particles.31 On the other hand, when strain-induced crystallization occurs, the molecular chains will spontaneously orient in the stretching direction and Δn will increase without applying additional stress. At the same time, the tensile stress tends to decrease.7, 16, 20, 32, 33, 34, 35, 36 Thus, the data point drifts to the upper-left direction with the progress of strain-induced crystallization.37 In some cases, these two types of deviations from the stress-optical law occur simultaneously, as shown shortly (see also Supplementary information).

Illustration for the interpretation of the Δn−σt diagram. Deformation of network chains is schematically drawn in this figure. The thick arrow from the origin to the upper-right direction indicates the Gaussian behavior that follows the stress-optical law.
For the presentation of general features of the experimental results, Figure 3 shows a set of diagrams for NR3 extended to αs=5 and kept for 1 h. During the extension process, σt and Δn increased in a seemingly similar manner (Figure 3a). While the specimen was subsequently held at the prefixed extension ratio, σt decreased with elapsed time, t (Figure 3b).14, 15 (The origin of t is taken at the moment when the extension is stopped.) This stress relaxation is mainly due to strain-induced crystallization because the molecular chains in crystals adopt the extended conformation aligned in the stretching direction.7, 16, 20, 32, 33, 34, 35, 36 We have already confirmed that the contribution of strain-induced crystallization to stress relaxation considerably outweighs that of disentanglement in the case of αs=6.14 As a result of strain-induced crystallization, Δn increased with time (Figure 3c). After 1 h, the sample was retracted to the original length, showing the lower σt (Figure 3a). The Δn−σt diagram during the extension and holding processes is shown in Figure 3d. At the beginning of extension, the Δn−σt relationship followed the stress-optical law. The slope of the linear plot near the origin, namely, the stress-optical coefficient (C), was 1.9 × 10−9 Pa−1, which was in good agreement with the results of earlier studies.13 This coincidence of C values indicates the reliability of the current experimental method on the basis of spectrum measurement, although a certain fluctuation of the estimated value was recognized. With the increase in extension ratio and σt, the data points slightly drifted to the right because of the finite extensibility of the network chains.30 (This trend is more clearly recognized in parts d and e of Figure 4.) During the 1 h of subsequent holding at αs=5, the data points gradually moved to the upper-left direction because of the progress of strain-induced crystallization as expected (Supplementary information). A similar effect of strain-induced crystallization forming a ‘hook’ in the Δn−σt diagram has been reported by Valladares et al.37 for vulcanized NR samples and by Ryu et al.30 for melted poly(ethylene terephthalate). On the other hand, the non-Gaussian behavior due to limited chain extensibility (namely, the drift of data points to the right in Δn−σt diagrams) has not been clearly recognized for NR vulcanizates in previous studies,22, 23, 24, 37, 38, 39 probably because the available ranges of Δn and σt were limited because of the occurrence of scattering by strain-induced crystals. Otherwise, the occurrence of strain-induced crystallization, which tends to drift the data points in the opposite direction, may have neutralized the deviation from the stress-optical law during slow deformation. The fast deformation rate and the birefringence measurement method in this study have enabled us to detect the peculiar behavior in the non-Gaussian region.

Results for NR3 extended to αs=5 and kept for 1 h. (a) Variation of σt and Δn with α during extension and contraction processes. Solid and broken lines indicate σt for the extension and contraction processes, respectively. Filled triangle indicates Δn for the extension process. (b) Stress relaxation with time at the fixed extension ratio. (c) Increase in Δn with time at the fixed extension ratio. (d) Δn−σt diagram during the extension process and the subsequent holding at the fixed extension ratio. The solid line near the origin indicates the regression line for the first three data points. The slope of this line is 1.92 × 10−9 Pa−1.

Δn−σt diagrams during the extension process and subsequent holding at the fixed extension ratio for the three NR samples. (a) αs=3, (b) αs=3.5, (c) αs=4. The inset is the enlargement near the origin. (d) αs=5, (e) αs=6.
According to the above-mentioned discussion, we have to be careful about the estimation of crystallinity of extended rubber samples on the basis of Δn and σt using the equation proposed by Taylor and Darin;40

where fc is the volume fraction of the crystalline parts, Δn0 is the birefringence of pure crystals and F is the orientation factor. This equation assumes the preservation of the stress-optical law, which is applicable only in the Gaussian region. In some cases, however, we cannot tell whether an extended and crystallized specimen is in the Gaussian region because of the simultaneous occurrence of the two types of deviations in the opposite direction from the stress-optical law (Figure 2). In such a case, the crystallinity estimated using Equation (7) would not be reliable; we may have to adopt the modified stress-optical law.29, 30
The Δn−σt diagrams for the three NR samples are summarized in Figure 4. In the case of αs⩽4, all three samples followed the stress-optical law during the extension process before ‘hooks’ were formed. When the samples were stretched more and σt exceeded ∼15 MP, the data points during the extension process tended to deviate from the stress-optical law (parts d and e of Figure 4). That is to say, the non-Gaussian behavior became pronounced with the increase in σt. This result suggests that the high tensile stress of cross-linked NR is related to finite chain extensibility, as has been discussed by Treloar,13 although contribution of strain-induced crystallization should not be ruled out.
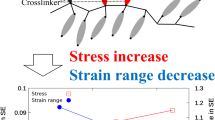
In Figure 4, the slope of the ‘hooks’ changed according to network-chain density and αs. In the case of αs=3 and 3.5 (parts a and b of Figure 4), the ‘hooks’ during the holding process sloped down to the left. In our previous studies using X-ray diffraction, we confirmed that our NR samples start crystallization at α=∼3.2 regardless of network-chain density.7 Therefore, strain-induced crystallization may have not occurred when αs=3. In the case of αs=3.5, although the strain ratio exceeded the incipient strain of crystallization, the growth rate of crystallites may have been very slow. (In Figure 1, I200 is very small at α=3.5.) Thus, no increase in Δn was observed during the holding (stress relaxation) process. On the other hand, when αs is 4 or larger, the ‘hooks’ basically sloped up to the left because of strain-induced crystallization. In addition, we can notice a trend that the sample with the lower network-chain density showed the steeper slope of the ‘hooks’. As an inversely extreme example, the increase in Δn for the ‘hook’ of NR1 in the case of αs=6 (Figure 4e) is almost negligible. Such a dependence of the slope of ‘hooks’ on network-chain density is explained as follows: the increase in Δn is brought about by the incorporation of unoriented chains into oriented crystals. That is to say, in the sample with the longer network-chain length (namely, the lower network-chain density) such as NR5, strain-induced crystals tend to incorporate unoriented chains from the surrounding amorphous phase. On the contrary, if highly oriented amorphous chains are converted to oriented crystals, Δn will not change much, in spite of the progress of strain-induced crystallization, because the degree of orientation in the specimen is almost maintained. This is the case for the highly stretched (αs=6) NR1. In this way, we can inspect using Δn−σt diagrams that the degree of extension of network chains to be incorporated into strain-induced crystals is quite different, depending on network-chain density and αs.
To compare the degree of stress relaxation during the holding process among samples having different moduli, the stress values normalized by maximum stress (hereafter, we refer to this normalized stress as fσ) should be evaluated.14, 15 Figure 5 shows the variation of normalized stress with time during the 1 h of holding. The abscissa is indicated by the logarithmic scale. In every case, fσ decreased almost linearly with the logarithm of elapsed time. At the same time, among samples with the same αs, plots almost overlap with each other. That is to say, fσ is a function of elapsed time (t) and αs, but is almost independent of network-chain density for the current samples (see also Figure 6). Although this peculiar feature has been reported in the case of αs=6,14, 15 the results for other αs values in this study newly demonstrate the generality of this feature. The final values of fσ after 1 h of holding are summarized in Figure 6. The acceleration of stress relaxation at the higher αs region is apparent, which should be attributable to strain-induced crystallization.14

Stress relaxation with time at a fixed extension ratio. (a) αs=3, (b) αs=3.5, (c) αs=4, (d) αs=5, (e) αs=6. In each part, the three plots almost overlap.

Degree of stress relaxation after 1 h of holding.
Here, we raise a question. According to our previous studies using X-ray diffraction, the rate of strain-induced crystallization depends on network-chain density.14, 15 If stress relaxation is accelerated by strain-induced crystallization, why is fσ independent of network-chain density (Figures 5 and 6)? Furthermore, the degree of extension of molecular chains to be incorporated into crystals depended on network-chain density, as has been discussed on the basis of Figure 4. Such a situation is also hard to connect to the behavior of fσ. This puzzle can be figured out if we adopt the two-component model19 with a slight modification. In the two-component model, the cross-linked rubber is regarded as a combination of an elastically effective component and a fluid-like one. Here, the elastically effective component is assumed to be responsible for rubber elasticity and for the onset of crystallization, whereas the fluid-like component is assumed to be ineffective for rubber elasticity. The physical properties of these two components are separately considered and apportioning to the observed quantity is discussed. For example, the fast and slow processes of stress relaxation are thought to be attributable to liquid-like and elastically effective components, respectively, according to our previous analysis.14 To explain the increase in Δn during strain-induced crystallization (Figure 4), we newly assume that the fluid-like component can be incorporated into existing strain-induced crystals. The elastically effective component in each sample is assumed to show the same crystallization behavior, because they should have the same mesh size in their swollen gels, reflecting the same composition of stearic acid and active zinc oxide (Table 1).41 Then the apparent value of network-chain density and tensile stress should reflect the fraction of the elastically effective component19 (Tosaka19 should be referred to for a further understanding of the two-component model).
The development of molecular orientation (viz., Δn) and stress relaxation during strain-induced crystallization is explained using the illustration in Figure 7, which is drawn on the basis of the two-component model. Figure 7a schematically shows the situation in extended samples before the onset of strain-induced crystallization. Overall tensile stress depends on the fraction of the elastically effective component (namely, the number of spring elements in this figure). Accordingly, the sample with the higher network-chain density shows the higher tensile stress. The elastically effective component should contain a considerable amount of highly stretched chains when the extension ratio is high. Then the effect of the finite chain extensibility, which shifts the data point in the Δn−σt diagram to the right (Figure 4) should be more pronounced for the sample with the higher network-chain density. On the other hand, statistical averaging of the degree of orientation among elastically effective components and fluid-like ones will result in Gaussian behavior for the sample with the lower network-chain density. When the extension ratio exceeds a certain critical value, the samples will subsequently crystallize (Figure 7b). Because the sample with the higher network-chain density contains the more elastically effective component, the increase in crystallinity for the whole sample is faster in the early stage.14, 15 On the other hand, fσ should depend on the degree of crystallization of only the elastically effective component (namely, on the fraction of the straight part in each of the spring elements).19 Because the crystallization rate of the elastically effective component in each sample should be the same, fσ may have shown the same behavior regardless of the different network-chain density (Figures 5 and 6).

Schematic illustration of stress relaxation and development of birefringence in the extended samples due to strain-induced crystallization on the basis of the two-component model. The spring elements drawn by solid lines represent the elastically effective component, whereas the winding broken lines represent the fluid-like component. (a) Before the onset of strain-induced crystallization. (b) After partial crystallization.
The trend found in the slopes of ‘hooks’ in the Δn−σt diagram (Figure 4) suggests the inclusion of the fluid-like component into the existing crystals. That is to say, the elastically effective component (spring elements) may crystallize first, functioning as nuclei. Subsequently, in the sample with the lower network-chain density, the larger fraction of isotropic amorphous chains (namely, the fluid-like component) is thought to be incorporated into strain-induced crystals (drawn by broken lines in Figure 7b), increasing the overall molecular orientation. Crystallization from the isotropic amorphous chains should favor the formation of lamellar crystals because of kinetics.42, 43 Accordingly, the shish-kebab structure with the shish and the kebab originating from elastically effective and fluid-like components, respectively, may be developed in this sample.7, 8 On the other hand, incorporation of the fluid-like component into strain-induced crystals should be negligible when Δn does not increase despite the development of strain-induced crystallization (e.g., NR1 in Figure 4e). As highly oriented amorphous chains should be directly converted into oriented crystals, microfibriller or fringed-micelle structures could be formed in such a sample.33, 44
The idea that the fluid-like component can be incorporated into existing crystals is also convenient for understanding the behavior of strain-induced crystallization during slow extension (Figure 1). In this case, once the incipient strain of crystallization is exceeded during the stretching process, the increase in crystallinity hardly depends on network-chain density.6, 8 If the elastically effective component is solely involved in strain-induced crystallization, a more rapid increase in crystallinity for the sample with the higher network-chain density is expected. We formerly thought that the discrepancy between this expectation and the experimental results comes from the modification of the network structure because of the formation of additional cross-linking points by the crystals. However, we now consider that the weak dependence of the increase in crystallinity on network-chain density is attributable to the inclusion of the more fluid-like component into strain-induced crystals for the sample with the lower network-chain density, reflecting the larger mobility of network chains.
Conclusion
Birefringence and tensile stress of vulcanized NR samples were measured simultaneously. The Δn−σt diagrams were interpreted in terms of the deviation from the stress-optical law because of the effect of finite chain extensibility and strain-induced crystallization. The non-Gaussian behavior in the Δn−σt diagrams could be clearly detected by virtue of the fast deformation rate and the birefringence measurement method adopted in this study; the non-Gaussian feature became pronounced with the increase in network-chain density and σt. At the same time, the formation of ‘hooks’ in the Δn−σt diagrams because of strain-induced crystallization was observed. The slopes of the ‘hooks’ suggested that, in the sample with the lower network-chain density, strain-induced crystals tended to incorporate unoriented chains from the surrounding amorphous phase, whereas highly oriented amorphous chains may have converted into oriented crystals in samples showing high tensile stress. The normalized stress decreased linearly with the logarithm of elapsed time in a manner that is almost independent of network-chain density. The two-component model19 could successfully explain these experimental results, assuming that the fluid-like component can be incorporated into existing strain-induced crystals.
References
Katz, J. R. Röntgenspektrographische Untersuchungen am gedehnten Kautschuk und ihre mögliche Bedeutung für das Problem der Dehnungseigenschaften dieser Substanz. Naturwissenschaften 19, 410–416 (1925).
Clark, G. L., LeTourneau, R. L. & Ball, J. M. Hysteresis in the crystallization of stretched vulcanized rubber. Rubber Chem. Technol. 14, 546–554 (1941).
Mitchell, G. R. A wide-angle X-ray study of the development of molecular orientation in crosslinked natural rubber. Polymer 25, 1562–1572 (1984).
Murakami, S., Senoo, K., Toki, S. & Kohjiya, S. Structural development of natural rubber during uniaxial stretching by in situ wide angle X-ray diffraction using a synchrotron radiation. Polymer 43, 2117–2120 (2002).
Toki, S., Sics, I., Ran, S., Liu, L., Hsiao, B. S., Murakami, S., Senoo, K. & Kohjiya, S. New insights into structural development in natural rubber during uniaxial deformation by in situ synchrotron X-ray diffraction. Macromolecules 35, 6578–6584 (2002).
Tosaka, M., Kohjiya, S., Murakami, S., Poompradub, S., Ikeda, Y., Toki, S., Sics, I. & Hsiao, B. S. Effect of network-chain length on strain-induced crystallization of NR and IR vulcanizates. Rubber Chem. Technol. 77, 711–723 (2004).
Tosaka, M., Murakami, S., Poompradub, S., Kohjiya, S., Ikeda, Y., Toki, S., Sics, I. & Hsiao, B. S. Orientation and crystallization of natural rubber network as revealed by WAXD using synchrotron radiation. Macromolecules 37, 3299–3309 (2004).
Tosaka, M. Strain-induced crystallization of crosslinked natural rubber as revealed by X-ray diffraction using synchrotron radiation. Polym. J. 39, 1207–1220 (2007).
Smith, K. J. Jr., Greene, A. & Ciferri, A. Crystallization under stress and non-Gaussian behavior of macromolecular networks. Kollid-Z. 194, 49–67 (1964).
Poompradub, S., Tosaka, M., Kohjiya, S., Ikeda, Y., Toki, S., Sics, I. & Hsiao, B. S. Mechanism of strain-induced crystallization in filled and unfilled natural rubber vulcanizates. J. Appl. Phys. 97, 103529/1–103529/9 (2005).
Trabelsi, S., Albouy, P.- A. & Rault, J. Crystallization and melting processes in vulcanized stretched natural rubber. Macromolecules 36, 7624–7639 (2003).
Chenal, J.- M., Chazeau, L., Guy, L., Bomal, Y. & Gauthier, C. Molecular weight between physical entanglements in natural rubber: A critical parameter during strain-induced crystallization. Polymer 48, 1042–1046 (2007).
Treloar, L. R. G. The Physics of Rubber Elasticity, 3rd edn. (Clarendon Press, Oxford, 1975).
Tosaka, M., Kawakami, D., Senoo, K., Kohjiya, S., Ikeda, Y., Toki, S. & Hsiao, B. S. Crystallization and stress relaxation in highly stretched samples of natural rubber and its synthetic analogue. Macromolecules 39, 5100–5105 (2006).
Tosaka, M., Senoo, K., Kohjiya, S. & Ikeda, Y. Crystallization of stretched network chains in crosslinked natural rubber. J. Appl. Phys. 101, 84909 (2007).
Flory, P. J. Thermodynamics of crystallization in high polymers. I. Crystallization induced by stretching. J. Chem. Phys. 15, 397–408 (1947).
Yamamoto, M. & White, J. L. Theory of deformation and strain-induced crystallization of an elastomeric network polymer. J. Polym. Sci., Part A-2 9, 1399–1415 (1971).
James, H. M. & Guth, E. Theory of the elastic properties of rubber. J. Chem. Phys. 11, 455–481 (1943).
Tosaka, M. A route for the thermodynamic description of strain-induced crystallization in sulfur-cured natural rubber. Macromolecules 42, 6166–6174 (2009).
Toki, S., Fujimaki, T. & Okuyama, M. Strain-induced crystallization of natural rubber as detected real-time by wide-angle X-ray diffraction technique. Polymer 41, 5423–5429 (2000).
Hashiyama, M., Gaylord, R. & Stein, R. S. Birefringence studies of the stress-induced crystallization of rubbers. Makromol. Chem., Suppl. 1, 579–597 (1975).
De Candia, F., Romano, G., Russo, R. & Vittoria, V. Optical and calorimetric analysis of stress-induced crystallization in natural rubber networks. J. Polym. Sci. Polym. Phys. Ed. 20, 1525–1531 (1982).
Chen, Y.- J., White, J. L., Min, K., Nakajima, N. & Weissert, F. C. A rheo-optical study of the influence of additives on crystallization rates in cis-1,4 polyisoprene. Rubber Chem. Technol. 61, 324–339 (1988).
Mott, P. H. & Roland, C. M. Elasticity of natural rubber networks. Macromolecules 29, 6941–6945 (1996).
Hongladarom, K., Burghardt, W. R., Baek, S. G., Cementwala, S. & Magda, J. J. Molecular alignment of polymer liquid crystals in shear flows. 1. Spectrographic birefringence technique, steady-state orientation, and normal stress behavior in poly(benzyl glutamate) solutions. Macromolecules 26, 772–784 (1993).
Medhat, M. & El-Zaiat, S. Y. Interferometric determination of the birefringence dispersion of anisotropic materials. Optics Commun. 141, 145–149 (1997).
Matsumoto, T. & Bogue, D. C. Stress birefringence in amorphous polymers under nonisothermal conditions. J. Polym. Sci., Polym. Phys. Ed. 15, 1663–1674 (1977).
Kröger, M., Luap, C. & Muller, R. Polymer melts under uniaxial elongational flow: Stress-optical behavior from experiments and nonequilibrium molecular dynamics computer simulations. Macromolecules 30, 526–539 (1997).
Inoue, T., Ryu, D.- S. & Osaki, K. A rheo-optical study on polystyrene under large tensile deformation around the glass transition temperature. Macromolecules 31, 6977–6983 (1998).
Ryu, D. S., Inoue, T. & Osaki, K. A birefringence study of polymer crystallization in the process of elongation of films. Polymer 39, 2515–2520 (1998).
Bokobza, L. & Erman, B. A Theoretical and experimental study of filler effect on stress-deformation-segmental orientation relations for poly(dimethylsiloxane) networks. Macromolecules 33, 8858–8864 (2000).
Miyamoto, Y., Yamao, H. & Sekimoto, K. Crystallization and melting of polyisoprene rubber under uniaxial deformation. Macromolecules 36, 6462–6471 (2003).
Toki, S., Sics, I., Ran, S., Liu, L., Hsiao, B. S., Murakami, S., Tosaka, M., Kohjiya, S., Poompradub, S., Ikeda, Y. & Tsou, A. H. Strain-induced molecular orientation and crystallization in natural rubber and synthetic rubbers under uniaxial deformation by in-situ synchrotron X-ray study. Rubber Chem. Technol. 77, 317–335 (2004).
Toki, S., Sics, I., Hsiao, B. S., Tosaka, M., Poompradub, S., Ikeda, Y. & Kohjiya, S. Probing the nature of strain-induced crystallization in polyisoprene rubber by combined thermomechanical and in situ X-ray diffraction techniques. Macromolecules 38, 7064–7073 (2005).
Trabelsi, S., Albouy, P.- A. & Rault, J. Stress-induced crystallization properties of natural and synthetic cis-polyisoprene. Rubber Chem. Technol. 77, 303–316 (2004).
Toki, S., Hsiao, B. S., Kohjiya, S., Tosaka, M., Tsou, A. H. & Datta, S. Synchrotron X-ray studies of vulcanized rubbers and thermoplastic elastomers. Rubber Chem. Technol. 79, 460–488 (2006).
Valladares, D., Toki, S., Sen, T. Z., Yalcin, B. & Cakmak, M. The effect of natural rubber crosslink density on real time birefringence, true stress and true strain behavior. Macromol. Symp. 185, 149–166 (2002).
Shimomura, Y., White, J. L. & Spruiell, J. E. A comparative study of stress-induced crystallization of Guayulee, Hevea and synthetic polyisoprenes. J. Appl. Polym. Sci. 27, 3553–3567 (1982).
Toki, S., Sen, T. Z., Valladares, D. & Cakmak, M. Proceeding of 159th Meeting of the Rubber Division, American Chemical Society, Providence, Rhode Island, April 24–27, Paper No. 12. (Rubber Division, American Chemical Society, Akron, 2001).
Taylor, G. R. & Darin, S. R. Birefringence and crystallization in elastomers. J. Appl. Phys. 26, 1075–1079 (1955).
Ikeda, Y., Higashitani, N., Hijikata, K., Kokubo, Y., Morita, Y., Shibayama, M., Osaka, N., Suzuki, T., Endo, H. & Kohjiya, S. Vulcanization: New focus on a traditional technology by small-angle neutron scattering. Macromolecules 42, 2741–2748 (2009).
Bassett, D. C. Principles of Polymer Morphology (Cambridge University Press, Cambridge, 1981).
Armistead, K., Goldbeck-Wood, G. & Keller, A. Polymer crystallization theories. Adv. Polym. Sci. 100, 221–312 (1992).
Toki, S. & Hsiao, B. S. Nature of strain-induced structures in natural and synthetic rubbers under stretching. Macromolecules 36, 5915–5917 (2003).
Acknowledgements
This study was partly supported by a Grant-in Aid for Scientific Research (C), No. 20550187, from the Japan Society for the Promotion of Science (to M Tosaka) and by the Heiwa Nakajima Foundation and by research grants from the President of Kyoto Institute of Technology (to Y Ikeda).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Polymer Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Tosaka, M., Kohjiya, S., Ikeda, Y. et al. Molecular orientation and stress relaxation during strain-induced crystallization of vulcanized natural rubber. Polym J 42, 474–481 (2010). https://doi.org/10.1038/pj.2010.22
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2010.22
Keywords
This article is cited by
-
Viscoelastic behavior with damage evolution of a new smart geosynthetic in service temperature range
Journal of Central South University (2022)
-
Thermally induced crystallization kinetics of uncrosslinked and unfilled synthetic cis-1,4-polyisoprene rubber monitored by shear rheological tests
Korea-Australia Rheology Journal (2016)
