
Abstract
Gene targeting by homologous recombination with exogenous DNA constructs is the most powerful technique available for analysis of mammalian gene function. Over the past several years, the methods used to generate knockout and knockin mice have been modified for use in cultured human cells. The most significant innovation has been the adaptation of recombinant adeno-associated viruses (rAAVs) for such targeting. The stages of rAAV-mediated gene targeting include (i) the design and construction of a DNA targeting vector, (ii) the production of an infectious rAAV stock, (iii) the generation of cell clones that harbor rAAV transgenes, (iv) screening for homologous recombinants and (v) the iterative targeting of multiple alleles. The protocol described herein allows the generation of a cell line with a single altered allele in 3 months. A second allele of the same gene can be targeted in an additional 3 months.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
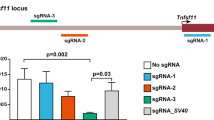
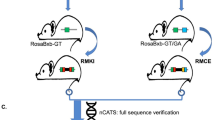
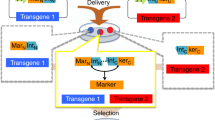
References
Bunz, F. Human cell knockouts. Curr. Opin. Oncol. 14, 73–78 (2002).
Cassman, M. Computational biology. Counting on the neuron. Science 300, 756–757 (2003).
Hannon, G.J. RNA interference. Nature 418, 244–251 (2002).
Sledz, C.A., Holko, M., de Veer, M.J., Silverman, R.H. & Williams, B.R. Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 5, 834–839 (2003).
Jackson, A.L. et al. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 21, 635–637 (2003).
Bridge, A.J., Pebernard, S., Ducraux, A., Nicoulaz, A.L. & Iggo, R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 34, 263–264 (2003).
Vogelstein, B. et al. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 319, 525–532 (1988).
Vogelstein, B. & Kinzler, K.W. The multistep nature of cancer. Trends Genet. 9, 138–141 (1993).
Wang, T.L. et al. Prevalence of somatic alterations in the colorectal cancer cell genome. Proc. Natl. Acad. Sci. USA 99, 3076–3080 (2002).
Sjoblom, T. et al. The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 (2006).
Lengauer, C., Kinzler, K.W. & Vogelstein, B. Genetic instability in colorectal cancers. Nature 386, 623–627 (1997).
Lengauer, C., Kinzler, K.W. & Vogelstein, B. Genetic instabilities in human cancers. Nature 396, 643–649 (1998).
Parsons, R. et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 75, 1227–1236 (1993).
Montrose-Rafizadeh, C. et al. Gene targeting of a CFTR allele in HT29 human epithelial cells. J. Cell Physiol. 170, 299–308 (1997).
Itzhaki, J.E., Gilbert, C.S. & Porter, A.C. Construction by gene targeting in human cells of a 'conditional' CDC2 mutant that rereplicates its DNA. Nat. Genet. 15, 258–265 (1997).
Hirata, R., Chamberlain, J., Dong, R. & Russell, D.W. Targeted transgene insertion into human chromosomes by adeno-associated virus vectors. Nat. Biotechnol. 20, 735–738 (2002).
Karakas, B. et al. Interleukin-1 alpha mediates the growth proliferative effects of transforming growth factor-beta in p21 null MCF-10A human mammary epithelial cells. Oncogene 25, 5561–5569 (2006).
Chamberlain, J.R. et al. Gene targeting in stem cells from individuals with osteogenesis imperfecta. Science 303, 1198–1201 (2004).
Kohli, M., Rago, C., Lengauer, C., Kinzler, K.W. & Vogelstein, B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 32, e3 (2004).
Cummins, J.M. et al. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 428 1 p following 486 (2004).
Sedivy, J.M. & Dutriaux, A. Gene targeting and somatic cell genetics—a rebirth or a coming of age? Trends Genet 15, 88–90 (1999).
Porteus, M.H. & Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 300, 763 (2003).
Yanez, R.J. & Porter, A.C. Therapeutic gene targeting. Gene Ther. 5, 149–159 (1998).
Topaloglu, O., Hurley, P.J., Yildirim, O., Civin, C.I. & Bunz, F. Improved methods for the generation of human gene knockout and knockin cell lines. Nucleic Acids Res. 33, e158 (2005).
Russell, D.W. & Hirata, R.K. Human gene targeting by viral vectors. Nat. Genet. 18, 325–330 (1998).
Choi, T., Huang, M., Gorman, C. & Jaenisch, R. A generic intron increases gene expression in transgenic mice. Mol. Cell. Biol. 11, 3070–3074 (1991).
Huang, M.T. & Gorman, C.M. Intervening sequences increase efficiency of RNA 3′ processing and accumulation of cytoplasmic RNA. Nucleic Acids Res. 18, 937–947 (1990).
Hellen, C.U. & Sarnow, P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 15, 1593–1612 (2001).
Park, B.H., Vogelstein, B. & Kinzler, K.W. Genetic disruption of PPARdelta decreases the tumorigenicity of human colon cancer cells. Proc. Natl. Acad. Sci. USA 98, 2598–2603 (2001).
Jallepalli, P.V. et al. Securin is required for chromosomal stability in human cells. Cell 105, 445–457 (2001).
Hanson, K.D. & Sedivy, J.M. Analysis of biological selections for high-efficiency gene targeting. Mol. Cell. Biol. 15, 45–51 (1995).
Friedel, R.H. et al. Gene targeting using a promoterless gene trap vector ('targeted trapping') is an efficient method to mutate a large fraction of genes. Proc. Natl. Acad. Sci. USA 102, 13188–13193 (2005).
Porteus, M.H., Cathomen, T., Weitzman, M.D. & Baltimore, D. Efficient gene targeting mediated by adeno-associated virus and DNA double-strand breaks. Mol. Cell. Biol. 23, 3558–3565 (2003).
Wilsker, D. & Bunz, F. Loss of ataxia telangiectasia mutated- and Rad3-related function potentiates the effects of chemotherapeutic drugs on cancer cell survival. Mol. Cancer Ther. 6, 1406–1413 (2007).
Wang, P., Yu, J. & Zhang, L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc. Natl. Acad. Sci. USA 104, 4054–4059 (2007).
Larochelle, S. et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 25, 839–850 (2007).
Kim, J.S., Lee, C., Bonifant, C.L., Ressom, H. & Waldman, T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol. Cell. Biol. 27, 662–677 (2007).
Hurley, P.J., Wilsker, D. & Bunz, F. Human cancer cells require ATR for cell cycle progression following exposure to ionizing radiation. Oncogene 26, 2535–2542 (2007).
Burkard, M.E. et al. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc. Natl. Acad. Sci. USA 104, 4383–4388 (2007).
Gallmeier, E. et al. Targeted disruption of FANCC and FANCG in human cancer provides a preclinical model for specific therapeutic options. Gastroenterology 130, 2145–2154 (2006).
Dang, L.H. et al. CDX2 has tumorigenic potential in the human colon cancer cell lines LOVO and SW48. Oncogene 25, 2264–2272 (2006).
Dang, D.T. et al. Hypoxia-inducible factor-1alpha promotes nonhypoxia-mediated proliferation in colon cancer cells and xenografts. Cancer Res. 66, 1684–1936 (2006).
Cunningham, S.C. et al. Targeted deletion of MKK4 in cancer cells: a detrimental phenotype manifests as decreased experimental metastasis and suggests a counterweight to the evolution of tumor-suppressor loss. Cancer Res. 66, 5560–5564 (2006).
Samuels, Y. et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 7, 561–573 (2005).
Papi, M., Berdougo, E., Randall, C.L., Ganguly, S. & Jallepalli, P.V. Multiple roles for separase auto-cleavage during the G2/M transition. Nat. Cell Biol. 7, 1029–1035 (2005).
Dang, D.T. et al. Glutathione I-transferase pi1 promotes tumorigenicity in HCT116 human colon cancer cells. Cancer Res. 65, 9485–9494 (2005).
Cummins, J.M. et al. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res. 64, 3006–3008 (2004).
Torrance, C.J., Agrawal, V., Vogelstein, B. & Kinzler, K.W. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat. Biotechnol. 19, 940–945 (2001).
Arena, S., Pisacane, A., Mazzone, M., Comoglio, P.M. & Bardelli, A. Genetic targeting of the kinase activity of the Met receptor in cancer cells. Proc. Natl. Acad. Sci. USA 104, 11412–11417 (2007).
Ho, S.N., Hunt, H.D., Horton, R.M., Pullen, J.K. & Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 (1989).
Dong, J.Y., Fan, P.D. & Frizzell, R.A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 7, 2101–2112 (1996).
Hirata, R.K. & Russell, D.W. Design and packaging of adeno-associated virus gene targeting vectors. J. Virol. 74, 4612–4620 (2000).
Vogelstein, B. & Gillespie, D. Preparative and analytical purification of DNA from agarose. Proc. Natl. Acad. Sci. USA 76, 615–619 (1979).
Vogelstein, B. Rapid purification of DNA from agarose gels by centrifugation through a disposable plastic column. Anal. Biochem. 160, 115–118 (1987).
Chan, T.A., Wang, Z., Dang, L.H., Vogelstein, B. & Kinzler, K.W. Targeted inactivation of CTNNB1 reveals unexpected effects of beta-catenin mutation. Proc. Natl. Acad. Sci. USA 99, 8265–8270 (2002).
Chan, T.A., Hwang, P.M., Hermeking, H., Kinzler, K.W. & Vogelstein, B. Cooperative effects of genes controlling the G(2)/M checkpoint. Genes Dev. 14, 1584–1588 (2000).
Chan, T.A., Hermeking, H., Lengauer, C., Kinzler, K.W. & Vogelstein, B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 401, 616–620 (1999).
Hwang, P.M. et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 7, 1111–1117 (2001).
Ramirez-Solis, R., Davis, A.C. & Bradley, A. Gene targeting in embryonic stem cells. Methods Enzymol. 225, 855–878 (1993).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rago, C., Vogelstein, B. & Bunz, F. Genetic knockouts and knockins in human somatic cells. Nat Protoc 2, 2734–2746 (2007). https://doi.org/10.1038/nprot.2007.408
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.408
This article is cited by
-
Common and mutation specific phenotypes of KRAS and BRAF mutations in colorectal cancer cells revealed by integrative -omics analysis
Journal of Experimental & Clinical Cancer Research (2021)
-
BAR-Seq clonal tracking of gene-edited cells
Nature Protocols (2021)
-
Generation and Characterization of a Skeletal Muscle Cell-Based Model Carrying One Single Gne Allele: Implications in Actin Dynamics
Molecular Neurobiology (2021)
-
Loss of SETDB1 decompacts the inactive X chromosome in part through reactivation of an enhancer in the IL1RAPL1 gene
Epigenetics & Chromatin (2018)
-
Mutant allele quantification reveals a genetic basis for TP53 mutation-driven castration resistance in prostate cancer cells
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
