
Abstract
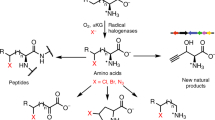
Enzymatic incorporation of a halogen atom is a common feature in the biosyntheses of more than 4,500 natural products1,2,3,4,5. Halogenation of unactivated carbon centers in the biosyntheses of several compounds of nonribosomal peptide origin is carried out by a class of mononuclear nonheme iron enzymes that require α-ketoglutarate (αKG, 1), chloride and oxygen6. To investigate the ability of these enzymes to functionalize unactivated methyl groups, we characterized the chlorination of the γ-methyl substituent of L-2-aminobutyric acid (L-Aba, 2) attached to the carrier protein CytC2 by iron halogenase (CytC3) from soil Streptomyces sp. We identified an intermediate state comprising two high-spin Fe(IV) complexes in rapid equilibrium. At least one of the Fe(IV) complexes abstracts hydrogen from the substrate. The demonstration that chlorination proceeds through an Fe(IV) intermediate that cleaves a C-H bond reveals the mechanistic similarity of aliphatic halogenases to the iron- and αKG-dependent hydroxylases.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
References
Gribble, G.W. Naturally occurring organohalogen compounds. Acc. Chem. Res. 31, 141–152 (1998).
van Pee, K.-H. & Unversucht, S. Biological dehalogenation and halogenation reactions. Chemosphere 52, 299–312 (2003).
Butler, A. & Carter-Franklin, J.N. The role of vanadium bromoperoxidase in the biosynthesis of halogenated marine natural products. Nat. Prod. Rep. 21, 180–188 (2004).
Gribble, G.W. Natural organohalogens: a new frontier for medicinal agents? J. Chem. Educ. 81, 1441–1449 (2004).
Fenical, W. & Jensen, P.R. Developing a new resource for drug discovery: marine actinomycete bacteria. Nat. Chem. Biol. 2, 666–673 (2006).
Vaillancourt, F.H., Yin, J. & Walsh, C.T. SyrB2 in syringomycin E biosynthesis is a nonheme FeII α-ketoglutarate- and O2-dependent halogenase. Proc. Natl. Acad. Sci. USA 102, 10111–10116 (2005).
Vaillancourt, F.H., Yeh, E., Vosburg, D.A., Garneau-Tsodikova, S. & Walsh, C.T. Nature's inventory of halogenation catalysts: oxidative strategies predominate. Chem. Rev. 106, 3364–3378 (2006).
Galonić, D.P., Vaillancourt, F.H. & Walsh, C.T. Halogenation of unactivated carbon centers in natural product biosynthesis: trichlorination of leucine during barbamide biosynthesis. J. Am. Chem. Soc. 128, 3900–3901 (2006).
Vaillancourt, F.H., Yeh, E., Vosburg, D.A., O'Connor, S.E. & Walsh, C.T. Cryptic chlorination by a non-haem iron enzyme during cyclopropyl amino acid biosynthesis. Nature 436, 1191–1194 (2005).
Ueki, M. et al. Enzymatic generation of the antimetabolite γ,γ-dichloroaminobutyrate by NRPS and mononuclear iron halogenase action in a Streptomycete. Chem. Biol. 13, 1183–1191 (2006).
Solomon, E.I. et al. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 100, 235–349 (2000).
Hausinger, R.P. Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 39, 21–68 (2004).
Costas, M., Mehn, M.P., Jensen, M.P. & Que, L., Jr. Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 104, 939–986 (2004).
Hanauske-Abel, H.M. & Günzler, V. A stereochemical concept for the catalytic mechanism of prolylhydroxylase. Applicability to classification and design of inhibitors. J. Theor. Biol. 94, 421–455 (1982).
Price, J.C., Barr, E.W., Tirupati, B., Bollinger, J.M., Jr. & Krebs, C. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 42, 7497–7508 (2003).
Price, J.C., Barr, E.W., Glass, T.E., Krebs, C. & Bollinger, J.M., Jr. Evidence for hydrogen abstraction from C1 of taurine by the high-spin Fe(IV) intermediate detected during oxygen activation by taurine:α-ketoglutarate dioxygenase (TauD). J. Am. Chem. Soc. 125, 13008–13009 (2003).
Bollinger, J.M., Jr. & Krebs, C. Stalking intermediates in oxygen activation by iron enzymes: motivation and method. J. Inorg. Biochem. 100, 586–605 (2006).
Proshlyakov, D.A., Henshaw, T.F., Monterosso, G.R., Ryle, M.J. & Hausinger, R.P. Direct detection of oxygen intermediates in the non-heme Fe enzyme taurine/α-ketoglutarate dioxygenase. J. Am. Chem. Soc. 126, 1022–1023 (2004).
Riggs-Gelasco, P.J. et al. EXAFS spectroscopic evidence for an Fe=O unit in the Fe(IV) intermediate observed during oxygen activation by taurine:α-ketoglutarate dioxygenase. J. Am. Chem. Soc. 126, 8108–8109 (2004).
Price, J.C., Barr, E.W., Hoffart, L.M., Krebs, C. & Bollinger, J.M., Jr. Kinetic dissection of the catalytic mechanism of taurine:α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 44, 8138–8147 (2005).
Hoffart, L.M., Barr, E.W., Guyer, R.B., Bollinger, J.M., Jr. & Krebs, C. Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc. Natl. Acad. Sci. USA 103, 14738–14743 (2006).
Blasiak, L.C., Vaillancourt, F.H., Walsh, C.T. & Drennan, C.L. Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature 440, 368–371 (2006).
Koehntop, K.D., Emerson, J.P. & Que, L., Jr. The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J. Biol. Inorg. Chem. 10, 87–93 (2005).
Quadri, L.E.N. et al. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 37, 1585–1595 (1998).
Pavel, E.G. et al. Circular dichroism and magnetic circular dichroism spectroscopic studies of the non-heme ferrous active site in clavaminate synthase and its interaction with α–ketoglutarate cosubstrate. J. Am. Chem. Soc. 120, 743–753 (1998).
Krebs, C. et al. Rapid freeze-quench 57Fe Mössbauer spectroscopy: monitoring changes of an iron-containing active site during a biochemical reaction. Inorg. Chem. 44, 742–757 (2005).
Pestovsky, O. et al. Aqueous FeIV=O: spectroscopic identification and oxo-group exchange. Angew. Chem. Int. Ed. 44, 6871–6874 (2005).
Ryle, M.J. et al. O2- and α-ketoglutarate-dependent tyrosyl radical formation in TauD, an α-keto acid-dependent non-heme iron dioxygenase. Biochemistry 42, 1854–1862 (2003).
Liu, P. et al. Oxygenase activity in the self-hydroxylation of (S)-2-hydroxypropylphosphonic acid epoxidase involved in fosfomycin biosynthesis. J. Am. Chem. Soc. 126, 10306–10312 (2004).
Neese, F. Theoretical spectroscopy of model-nonheme [Fe(IV)OL5]2+ complexes in their lowest triplet and quintet states using multireference ab initio and density functional theory methods. J. Inorg. Biochem. 100, 716–726 (2006).
Kojima, T., Leising, R.A., Yan, S. & Que, L., Jr . Alkane functionalization at nonheme iron centers. Stoichiometric transfer of metal-bound ligands to alkane. J. Am. Chem. Soc. 115, 11328–11335 (1993).
Burzlaff, N.I. et al. The reaction cycle of isopenicillin N synthase observed by X-ray diffraction. Nature 401, 721–724 (1999).
Acknowledgements
E.R. Strieter is gratefully acknowledged for a generous gift of (R,R)-(−)-pseudoephedrine glycinamide hydrate. We thank F.H. Vaillancourt and J.A. Read for careful proofreading of the manuscript. This work was supported by the US National Institutes of Health (NIH GM-69657 to J.M.B. and C.K.; GM-20011 and GM-49338 to C.T.W.), the donors of the American Chemical Society Petroleum Research Fund (ACS-PRF 41170-G3 to C.K.), the Arnold and Mabel Beckman Foundation (Young Investigator Award to C.K.) and the Dreyfus Foundation (Camille Dreyfus Teacher Scholar Award to C.K.). D.P.G. is supported by the Damon Runyon Cancer Research Foundation Postdoctoral Fellowship (DRG-1893-05).
Author information
Authors and Affiliations
Contributions
D.P.G., C.T.W., J.M.B. and C.K. designed the experiments and wrote the manuscript. D.P.G. overproduced and purified proteins, synthesized deuterated substrate, and performed iron titration and SF absorption experiments. D.P.G. and E.W.B. performed FQ experiments. C.K. collected and analyzed Mössbauer spectra.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Fig. 1
Comparison of the 4.2 K/zero-field Mössbauer reference spectrum of the Fe(IV) intermediates as described in the main manuscript (solid lines) to reference spectra of the Fe(IV) intermediates of samples prepared under modified reaction conditions. (PDF 108 kb)
Supplementary Scheme 1
Enzymatic conversion of L-Aba to γ-Cl-L-Aba in cytotrienin-producing Streptomyces sp. (PDF 66 kb)
Rights and permissions
About this article
Cite this article
Galonić, D., Barr, E., Walsh, C. et al. Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat Chem Biol 3, 113–116 (2007). https://doi.org/10.1038/nchembio856
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nchembio856
This article is cited by
-
A theoretical study for spin-dependent hydrogen abstraction by non-heme FeIVO complexes based on DFT potential energy surfaces
Theoretical Chemistry Accounts (2023)
-
Algorithm-aided engineering of aliphatic halogenase WelO5* for the asymmetric late-stage functionalization of soraphens
Nature Communications (2022)
-
Reaction pathway engineering converts a radical hydroxylase into a halogenase
Nature Chemical Biology (2022)
-
The chloroalkaloid (−)-acutumine is biosynthesized via a Fe(II)- and 2-oxoglutarate-dependent halogenase in Menispermaceae plants
Nature Communications (2020)
-
The oxidation of cyclo-olefin by the S = 2 ground-state complex [FeIV(O)(TQA)(NCMe)]2+
JBIC Journal of Biological Inorganic Chemistry (2020)
