
Abstract
RNA interference (RNAi) is the process of sequence-specific, post-transcriptional gene silencing in animals and plants, initiated by double-stranded RNA (dsRNA) that is homologous in sequence to the silenced gene1,2,3,4. The mediators of sequence-specific messenger RNA degradation are 21- and 22-nucleotide small interfering RNAs (siRNAs) generated by ribonuclease III cleavage from longer dsRNAs5,6,7,8,9. Here we show that 21-nucleotide siRNA duplexes specifically suppress expression of endogenous and heterologous genes in different mammalian cell lines, including human embryonic kidney (293) and HeLa cells. Therefore, 21-nucleotide siRNA duplexes provide a new tool for studying gene function in mammalian cells and may eventually be used as gene-specific therapeutics.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
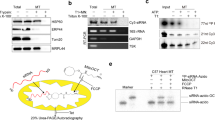
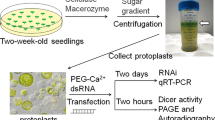

References
Fire, A. RNA-triggered gene silencing. Trends Genet. 15, 358–363 (1999).
Sharp, P. A. RNA interference 2001. Genes Dev. 15, 485–490 (2001).
Hammond, S. M., Caudy, A. A. & Hannon, G. J. Post-transcriptional gene silencing by double-stranded RNA. Nature Rev. Genet. 2, 110–1119 (2001).
Tuschl, T. RNA interference and small interfering RNAs. Chem. Biochem. 2, 239–245 (2001).
Hamilton, A. J. & Baulcombe, D. C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286, 950–952 (1999).
Hammond, S. M., Bernstein, E., Beach, D. & Hannon, G. J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404, 293–296 (2000).
Zamore, P. D., Tuschl, T., Sharp, P. A. & Bartel, D. P. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101, 25–33 (2000).
Bernstein, E., Caudy, A. A., Hammond, S. M. & Hannon, G. J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366 (2001).
Elbashir, S. M., Lendeckel, W. & Tuschl, T. RNA interference is mediated by 21 and 22 nt RNAs. Genes Dev. 15, 188–200 (2001).
Caplen, N. J., Fleenor, J., Fire, A. & Morgan, R. A. dsRNA-mediated gene silencing in cultured Drosophila cells: a tissue culture model for the analysis of RNA interference. Gene 252, 95–105 (2000).
Clemens, J. C. et al. Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc. Natl Acad. Sci. USA 97, 6499–6503 (2000).
Ui-Tei, K., Zenno, S., Miyata, Y. & Saigo, K. Sensitive assay of RNA interference in Drosophila and Chinese hamster cultured cells using firefly luciferase gene as target. FEBS Lett. 479, 79–82 (2000).
Wianny, F. & Zernicka-Goetz, M. Specific interference with gene function by double-stranded RNA in early mouse development. Nature Cell Biol. 2, 70–75 (2000).
Svoboda, P., Stein, P., Hayashi, H. & Schultz, R. M. Selective reduction of dormant maternal mRNAs in mouse oocytes by RNA interference. Development 127, 4147–4156 (2000).
Bahramian, M. B. & Zarbl, H. Transcriptional and posttranscriptional silencing of rodent alpha(I) collagen by a homologous transcriptionally self-silenced transgene. Mol. Cell. Biol. 19, 274–283 (1999).
Stark, G. R., Kerr, I. M., Williams, B. R., Silverman, R. H. & Schreiber, R. D. How cells respond to interferons. Annu. Rev. Biochem. 67, 227–264 (1998).
Manche, L., Green, S. R., Schmedt, C. & Mathews, M. B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell. Biol. 12, 5238–5248 (1992).
Minks, M. A., West, D. K., Benvin, S. & Baglioni, C. Structural requirements of double-stranded RNA for the activation of 2′,5′-oligo(A) polymerase and protein kinase of interferon-treated HeLa cells. J. Biol. Chem. 254, 10180–10183 (1979).
Clemens, M. & Williams, B. Inhibition of cell-free protein synthesis by pppA2′p5′A2′p5′A: a novel oligonucleotide synthesized by interferon-treated L cell extracts. Cell 13, 565–572 (1978).
Macejak, D. G. et al. Inhibition of hepatitis C virus (HCV)-RNA-dependent translation and replication of a chimeric HCV poliovirus using synthetic stabilized ribozymes. Hepatology 31, 769–776 (2000).
Kehlenbach, R. H., Dickmanns, A. & Gerace, L. Nucleocytoplasmic shuttling factors including Ran and CRM1 mediate nuclear export of NFAT In vitro. J. Cell. Biol. 141, 863–874 (1998).
Kreis, T. & Vale, R. Guidebook to the Cytoskeletal and Motor Proteins, Parts 2b and 3a (Oxford Univ. Press, Oxford, 1999).
Sullivan, T. et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147, 913–920 (1999).
Wassenegger, M. RNA-directed DNA methylation. Plant Mol. Biol. 43, 203–220 (2000).
Mette, M. F., Aufsatz, W., van der Winden, J., Matzke, M. A. & Matzke, A. J. M. Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J. 19, 5194–5201 (2000).
Wang, M.-B., Wesley, S. V., Finnegan, E. J., Smith, N. A. & Waterhouse, P. M. Replicating satellite RNA induces sequence-specific DNA methylation and truncated transcripts in plants. RNA 7, 16–28 (2001).
Razin, A. CpG methylation, chromatin structure and gene silencing—a three-way connection. EMBO J. 17, 4905–4908 (1998).
Röber, R. A., Gieseler, R. K., Peters, J. H., Weber, K. & Osborn, M. Induction of nuclear lamins A/C in macrophages in in vitro cultures of rat bone marrow precursor cells and human blood monocytes, and in macrophages elicited in vivo by thioglycollate stimulation. Exp. Cell Res. 190, 185–194 (1990).
Harborth, J., Wang, J., Gueth-Hallonet, C., Weber, K. & Osborn, M. Self assembly of NuMA: multiarm oligomers as structural units of a nuclear lattice. EMBO J. 18, 1689–1700 (1999).
Parrish, S., Fleenor, J., Xu, S., Mello, C. & Fire, A. Functional anatomy of a dsRNA trigger: Differential requirement for the two trigger strands in RNA Interference. Mol. Cell 6, 1077–1087 (2000).
Acknowledgements
We thank J. Martinez, J. Ludwig and D. Bartel for comments on the manuscript; L. Fredel for help with image processing; H.-J. Dehne for technical assistance; F. Döring, R. Nehring, D. Ingelfinger and C. Schneider for supplying cell lines; A. Dickmanns for the gift of the plasmid pAD3; and R. Lührmann for support. This work was funded by a Biofuture grant of the Bundesministerium für Bildung und Forschung.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Elbashir, S., Harborth, J., Lendeckel, W. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001). https://doi.org/10.1038/35078107
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/35078107
This article is cited by
-
MicroRNAs and long non-coding RNAs during transcriptional regulation and latency of HIV and HTLV
Retrovirology (2024)
-
NKG2D knockdown improves hypoxic-ischemic brain damage by inhibiting neuroinflammation in neonatal mice
Scientific Reports (2024)
-
RNA interference in the era of nucleic acid therapeutics
Nature Biotechnology (2024)
-
Small interfering RNA (siRNA) as a potential gene silencing strategy for diabetes and associated complications: challenges and future perspectives
Journal of Diabetes & Metabolic Disorders (2024)
-
Hypoxia-driven deSUMOylation of EXOSC10 promotes adaptive changes in the transcriptome profile
Cellular and Molecular Life Sciences (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
