
Abstract
Brenner tumors are ovarian tumors, usually benign, containing epithelium that resembles transitional epithelium. As with other epithelial tumors there exist frankly malignant tumors and tumors that display greater proliferation than the benign Brenner tumors but lack destructive infiltrative growth, and these have been designated ‘atypical proliferative’ (borderline) Brenner tumors. There have been no well-documented cases of atypical proliferative Brenner tumors that have exhibited malignant behavior. Based on shared morphologic features it is generally believed that atypical proliferative Brenner tumors develop from benign Brenner tumors. The aim of the present study was to confirm this impression by investigating the immunohistochemical and molecular genetic features of benign and atypical proliferative Brenner tumors. Immunohistochemical staining for p16, fluorescence in-situ hybridization (FISH) for CDKN2A (p16-encoding gene) and mutational analysis of the genes commonly mutated in ovarian tumors were performed. p16 immunostaining was positive in the epithelial component of 12 (92%) of 13 benign Brenner tumors, but completely negative in all 7 atypical proliferative Brenner tumors. FISH identified homozygous deletion of CDKN2A in the epithelial component of all atypical proliferative Brenner tumors, but CDKN2A was retained in all benign Brenner tumors. Two PIK3CA somatic mutations were detected in the stromal component in 1 (5%) of 20 Brenner tumors and 3 somatic mutations (1 in KRAS and 2 in PIK3CA) were identified in the atypical epithelial component of 2 (29%) of 7 atypical proliferative Brenner tumors. In summary, our findings suggest that loss of CDKN2A and, to a lesser extent, KRAS and PIK3CA somatic mutations have a role in the progression of a benign to an atypical proliferative Brenner tumor.
Similar content being viewed by others
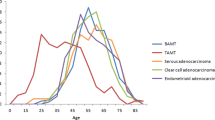

Main
The World Health Organization classification divides Brenner tumors into three categories: benign, borderline (atypical proliferative), and malignant.1 Benign Brenner tumors are composed of nests of mature transitional-like epithelial cells, which are surrounded by an abundant fibromatous stroma. Atypical proliferative Brenner tumors are characterized by large, irregularly shaped nests, and masses of similar appearing epithelium surrounded by dense fibromatous tissue. Some are composed of complex fibrovascular papillae, protruding into cystic spaces. Atypical proliferative Brenner tumors are usually associated with a benign Brenner tumor. Malignant Brenner tumors are characterized by cytologic atypia and stromal invasion in a context of a benign or atypical proliferative Brenner tumor.
We recently investigated the morphologic features and the immunohistochemical profile of the ‘tuboperitoneal junction’, which is the junction between the epithelium of the fimbria and the tubal mesothelial serosa, and proposed that it might be a ‘hot spot’ for ovarian carcinogenesis, and a possible site of origin of Brenner tumors.2 A subsequent study provided evidence, suggesting a tubal origin of Brenner tumors through transitional metaplasia and Walthard cell nests, based on their anatomical proximity, similar immunohistochemical profile, and the presence of cilia. In addition, we showed that luteinized stromal cells immediately adjacent to the epithelial nests, as well as the epithelial nests themselves, are involved in androgen biosynthesis in view of the presence of AKR1C3, a key enzyme in the production of androgens, in these cells and that hormone stimulation therefore probably has a role in the development of Brenner tumors as the epithelial component contains androgen receptor.3 The aim of the present study was to evaluate the immunohistochemical and molecular genetic features of benign and atypical proliferative Brenner tumors, and to elucidate their relationship and identify molecular alterations that underlie progression to atypical proliferative Brenner tumors.
Materials and methods
Case Selection
A total of 20 benign Brenner tumors and 7 atypical proliferative Brenner tumors were retrieved from the pathology files of the Johns Hopkins Hospital (Baltimore, MD), Washington Hospital Center (Washington, DC), and Seirei Mikatahara Hospital (Hamamatsu, Japan). All available slides were reviewed and clinicopathological data collected. This study was approved by the Institutional Review Board of the Johns Hopkins Medical Institutions.
Immunohistochemistry
Immunohistochemistry was performed using antibody against p16 (Ventana, cat# 705-4713, Ventana Medical Systems, Tucson, AZ, USA). Formalin-fixed paraffin-embedded sections were immunostained automatically in Benchmark XTMD (Ventana Medical Systems) using a previously described protocol.4, 5, 6 In summary, 4-μm-thick tissue sections were deparaffinized in xylene, and rehydrated in graded alcohols. Antigen unmasking was carried out by incubating the slides in Target retrieval solution (cat# S1699, Dako, Carpinteria, CA, USA) in a steamer for 30 min. EnVision™+/HRP, Rabbit polymer (cat# K4003, Dako) was applied for 30 min followed by 5 min of incubation with streptavidin peroxidase (Dako Liquid DAB+ cat# K3468, Dako). The slides were counterstained with hematoxylin for 3 min (cat# HHS32, Sigma-Aldrich, St Louis, MO, USA), dehydrated in graded ethanols and xylene. For a negative control, the primary antibody was replaced with TBS-t.
A positive reaction was defined as diffuse staining in at least 70% of the cells (diffuse), multifocal positivity in at least 20% of the cells (patchy), or scattered cells in less than 20% of the cells (focal). In addition, subcellular localization (nuclear or cytoplasmic) was recorded, and all observations were made separately for both epithelial and stromal components.
Fluorescence In-Situ Hybridization (FISH)
CDKN2A copy number per cell was evaluated using a two-color FISH assay. Briefly, 4-μm-thick sections were incubated for 30 min at 62 °C, deparaffinized in xylene, hydrated through graded ethanols, and placed in deionized water. The sections were incubated with proteinase K for 30 min at 37 °C and then washed in 2 × Aniara saline-sodium citrate (SSC). The slides were then placed in a denaturation solution (70% formamide/2 × SCC) for 5 min at 75 °C, rinsed in 2 × SSC, and allowed to cool at room temperature for 5 min. The slides were then dehydrated through graded ethanols and dried in an oven at 62 °C for 2 min. Vysis LSI CDKN2A SpectrumOrange/CEP9 SpectrumGreen probes (cat# 05J51-001, Abbott Molecular, IL, USA) were applied to the slides according to the vendor’s instructions and then coverslipped. Denaturation was accomplished by incubating the slides for 10 min at 80 °C. Hybridization was performed at 37 °C for 20–24 h. Coverslips were carefully removed and the slides were washed for 20 min in 1.5 mol/l urea in 0.2 × SSC, followed by washing in 2 × SSC for 2 min at room temperature. The slides were drained, dehydrated through graded ethanols, air-dried, mounted with ProLong® Gold Antifade Reagent with DAPI (cat# P-36931, Invitrogen, Eugene, OR, USA) and imaged.7 The CDKN2A copy number was assessed in at least 100 nuclei. Cases lacking both CDKN2A signals in the presence of CEP9 probe signal in >20% nuclei were considered homozygously deleted.
Tissue Microdissection and DNA Extraction
In order to extract DNA from the epithelial and stromal components separately, serial 10-μm formalin-fixed paraffin-embedded sections were placed onto UV-treated PALM membrane slides (Carl Zeiss MicroImaging GmbH, München, Germany) and stained with hematoxylin. Approximately 1000–2000 cells from the nests of transitional-type epithelium from Brenner tumors and atypical proliferative Brenner tumors were laser-capture microdissected using PALM Micro Beam System (Carl Zeiss MicroImaging). The stromal tissue surrounding the nests of epithelium was manually microdissected as previously described.5 Genomic DNA was extracted for sequencing using a QIAamp DNA Micro Kit (Qiagen, Valencia, CA, USA) according to the vendor’s protocol.
PCR Amplification and Mutation Analysis
Mutation analysis was performed for BRAF (exon 15), CTNNB1 (β-catenin-encoding gene, exon 3), ERBB2 (exon 20), FOXL2 (exon 1), KRAS (exon 2), PIK3CA (exons 1, 9, and 20), and PPP2R1A (exons 5 and 6), which are commonly mutated in ovarian tumors.8, 9 These exons were PCR amplified using primers shown in Table 1. All PCR amplification products were visualized with ethidium bromide under ultraviolet light by electrophoresis in 2.5% agarose gel. All amplified PCR products were sequenced at Agencourt Biosciences (Beverly, MA, USA) and all sequence variants were confirmed on at least three independent rounds.
Statistical Analysis
Comparisons of immunohistochemistry and FISH outcomes were performed using the two-tailed unpaired Fisher’s exact test. P-values of 0.05 or less were considered statistically significant. Statistical analysis was carried out using GraphPad Prism software version 5.0 (GraphPad Software, San Diego, CA, USA).
Results
p16 Immunostaining and CDKN2A FISH Assay
p16 immunohistochemical staining has been shown to be frequently negative in Brenner tumors.10, 11 We performed p16 immunostaining and CDKN2A, gene encoding both p16 (INK4a) and p14 (ARF), FISH assay on 13 Brenner tumors and 7 atypical proliferative Brenner tumors in order to evaluate whether they demonstrated p16 alterations.
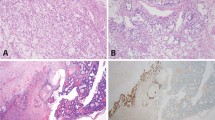
All benign Brenner tumors expressed p16 in the epithelial component, either in a diffuse or patchy distribution (Figure 1), except in one case which was negative (Table 2). In contrast, all seven atypical proliferative Brenner tumors were negative for p16 in the epithelial component (Table 2). Interestingly, five atypical proliferative Brenner tumors with an associated benign Brenner tumor showed loss of p16 expression in the benign epithelial component, but in two of these cases focal positivity restricted to few epithelial nests was noted (Figure 2).

Representative images of histological appearance (hematoxylin–eosin, HE), p16 immunostaining, and fluorescence in-situ hybridization for CDKN2A of benign Brenner tumor (BT) and atypical proliferative Brenner tumor (APBT). Benign BT shows intense and diffuse epithelial p16 positivity, both cytoplasmic and nuclear, and normal copy number (either 1 or 2) for CDKN2A (red) as compared with centromere region (CEP9, green) in both epithelial cells (filled arrow) and stromal cells (empty arrow). APBT is consistently negative for p16 and shows complete loss of CDKN2A (red) and retention of centromere 9 (CEP9, green) in the majority of epithelial cells (filled arrow) as compared with stromal cells, where both signals are identified in normal number (empty arrow). Higher magnification in the insets.

Histological appearance (hematoxylin–eosin, HE), p16 immunostaining, and fluorescence in-situ hybridization for CDKN2A of benign Brenner component in the context of atypical proliferative Brenner tumor. In the upper panels, the benign Brenner tumor component does not express p16, and shows complete loss of CDKN2A signals (red) and retention of centromere 9 signals (CEP9, green) in the majority of epithelial cells (filled arrow) as compared with stromal cells, where both signals are identified in normal number (empty arrow). In the lower panels, the benign Brenner tumor component associated with the atypical proliferative Brenner tumor shows focal p16 positivity, limited to one epithelial nest, retention of CDKN2A signals (red), and centromere signals (green; empty arrow), and adjacent p16 negative atypical proliferative Brenner tumor component, with loss of CDKN2A signals (red; filled arrow). Higher magnification in the insets.
CDKN2A FISH was performed to investigate CDKN2A copy number and its correlation with p16 immunostaining. The results are summarized in Table 2. None of the 13 Brenner tumors showed CDKN2A copy number alterations, in either the epithelial or stromal components (Figure 1). All seven atypical proliferative Brenner tumors showed CDKN2A homozygous deletion (Figure 1). Interestingly, the benign Brenner tumor component associated with five atypical proliferative Brenner tumors also showed mainly homozygous loss (Figure 2). In addition, in two of these cases, the benign Brenner tumor focally retained CDKN2A (Figure 2). CDKN2A copy number aberrations were not identified in the stromal component. Thus, there was a strong correlation between p16 immunohistochemical expression and CDKN2A copy number, and between CDKN2A copy number and tumor type (P-value <0.0001, two-tailed Fisher’s exact test, Table 3).
Mutational Analysis
A total of 20 Brenner tumors and 7 atypical proliferative Brenner tumors were analyzed for mutations of BRAF, CTNNB1 (β-catenin-encoding gene), ERBB2, FOXL2, KRAS, PIK3CA, and PPP2R1A genes, which are commonly mutated in ovarian tumors.8, 9 The transitional-type epithelium and the stromal component were analyzed separately. Two PIK3CA mutations were identified in the stromal component in one (5%) of 20 benign Brenner tumors, but not in the epithelial component, confirming the somatic nature of the mutation (Table 4 and Figure 3). The mutations identified in this benign Brenner tumor were a point mutation in position c.1634A>C that results in an amino-acid substitution at position 545 from a glutamic acid (E) to an alanine (A), and a single-nucleotide deletion of G at position c.1658 causing a frameshift mutation. None of the other benign Brenner tumors harbored mutations in the genes investigated.

Chromatograms and annotations show PIK3CA somatic mutations in the stromal component (below) of one benign Brenner tumor (BT11) compared with the wild-type sequences of the epithelial component (above). Chromatograms and annotations of the three somatic mutations identified in the epithelial component (below) of two atypical proliferative Brenner tumors (APBT2 and APBT6), two in PIK3CA, and one in KRAS, compared with the wild-type sequence of the stromal component (above).
In two (29%) of seven atypical proliferative Brenner tumors (APBT2 and APBT6), three somatic mutations, one in KRAS and two in PIK3CA, were identified in the atypical epithelial component but not in the stromal component (Table 4 and Figure 3). Interestingly, the benign component associated with APBT6 did not harbor the two mutations in KRAS and PIK3CA identified in the atypical component. APBT2 harbored a point mutation in position c.310C>A causing an amino-acid substitution at position 104 from a proline (P) to threonine (T). APBT6 harbored a KRAS point mutation c.35G>A, leading to an amino-acid substitution at position 12 from a glycine (G) to an aspartic acid (D), and a PIK3CA point mutation c.35472A>G resulting in an amino-acid substitution at position 1047 from histidine (H) to arginine (R).
Discussion
Atypical proliferative Brenner tumors are composed of large, irregularly shaped masses of transitional-type epithelium surrounded by dense fibromatous tissue, which usually are associated with a benign Brenner tumor component. The morphological similarity and common association of these two tumors suggest atypical proliferative Brenner tumors develop from benign Brenner tumors.1 The present study was undertaken in order to investigate molecular genetic characteristics of Brenner tumors focusing on the events that might underlie progression to atypical proliferative Brenner tumors.
CDKN2A, the gene encoding both p16 (INK4a) and p14 (ARF), is a tumor-suppressor gene frequently inactivated in human neoplasms by promoter hypermethylation and homozygous deletion.12 In the present study, we identified CDKN2A loss in all atypical proliferative Brenner tumors, whereas it was retained in all the benign Brenner tumors. Moreover, in two of the five atypical proliferative Brenner tumors that were associated with a benign Brenner tumor, CDKN2A was lost with just focal retention in the benign Brenner tumor. We found strong correlation between p16 immunohistochemical expression and CDKN2A copy number, so that p16 expression in the epithelial component of Brenner tumors was associated with retention of CDKN2A, whereas negative p16 immunostaining was associated with a complete loss of CDKN2A. Finally, there was correlation between CDKN2A copy number and tumor types, all benign Brenner tumors retained CDKN2A, whereas all atypical proliferative Brenner tumors lost this gene. Taken together, these findings suggest that loss of CDKN2A is involved in the progression from a benign to an atypical proliferative Brenner tumor. Accordingly, loss of expression of p16 by immunohistochemistry can be useful in distinguishing a benign Brenner tumor from an atypical proliferative Brenner tumor, in difficult cases. Consistent with our findings, two previous studies found that there was loss of p16 expression in Brenner tumors.10, 11 In particular, Cuatrecasas et al. reported low immunohistochemical expression or absence of p16 in benign, atypical proliferative, and malignant Brenner tumors.10 In addition, this study found loss of heterozygosity and promoter hypermethylation at the p16 locus in one malignant Brenner tumor. In the second study, Ali et al. reported p16 immunohistochemical expression in three benign Brenner tumors, and absence of p16 in three atypical proliferative and one malignant Brenner tumors.11 In any case, to our knowledge, ours is the first study that consistently identified homozygous deletion of CDKN2A, strongly correlated with loss of immunohistochemical expression of p16, in all atypical proliferative Brenner tumors examined, and CDKN2A retention, with associated p16 immunopositivity, in all benign Brenner tumors. Finally, CDKN2A has been shown to be frequently lost in ovarian mucinous tumors by SNP array.13, 14 The transitional epithelium of Brenner tumor frequently shows focal mucinous differentiation, and mucinous tumors (cystadenomas or atypical proliferative tumors) are occasionally identified concurrently with a Brenner tumor.1 The common loss of CDKN2A in both tumors provides additional molecular evidence showing their close association.
The identification of two PIK3CA somatic mutations in the stromal component of a single (5%) benign Brenner tumor suggests that mutations of the genes analyzed are unlikely to be common driver events underlying the development of these tumors. Nonetheless, two (29%) of seven atypical proliferative Brenner tumors harbored somatic mutation of KRAS and/or PIK3CA, neither of which was detected in the associated benign Brenner tumor, when present. Hence, these findings suggest a possible role of KRAS and PIK3CA somatic mutations in the progression from a benign to an atypical proliferative Brenner tumor. E545A and H1047R mutations of PIK3CA occur within the helical and kinase domain, respectively, and both have increased catalytic activity enhancing downstream signaling and oncogenic transformation in vitro.16 To our knowledge, the only previous study, which investigated somatic mutations in Brenner tumors, identified PIK3CA mutations in one (33%) of three atypical proliferative Brenner tumors, but did not interrogate the concurrent benign component.10 The mutations identified in this study have been previously described in other ovarian tumors.8, 17 For example, PIK3CA is also mutated in 5–45% ovarian endometrioid, clear cell, and low-grade serous carcinomas.8, 17 Finally, it is not surprising that KRAS mutation, the most common mutation identified in ovarian mucinous tumors, is identified in a minority of atypical proliferative Brenner tumors, as noted above they are frequently associated and the majority of Brenner tumors shows mucinous differentiation.3
In conclusion, our current study confirms that Brenner tumor can be considered a type I ovarian tumor based on the slow biological progression and stepwise acquisition of molecular genetic alterations.8, 18 In particular, it appears that CDKN2A loss, and to a lesser extent KRAS and PIK3CA mutations, may have a role in the progression of a benign Brenner tumor to an atypical proliferative Brenner tumor.
References
Lee KR, Tavassoli FA, Prat J et al. Surface epithelial-stromal tumours In: Tavassoli FA, Devilee P (eds). Pathology and Genetics: Tumours of the Breast and Female Genital Organs. World Health Organization Classification of Tumours 2003. IARC Press: Lyon, 2003, pp 140–143.
Seidman JD, Yemelyanova A, Zaino RJ et al. The fallopian tube-peritoneal junction: a potential site of carcinogenesis. Int J Gynecol Pathol 2011;30:4–11.
Kuhn E, Ayhan A, IeM Shih et alOvarian Brenner tumor. A morphologic, and immunohistochemical analysis suggesting an origin from fallopian tube epithelium (submitted).
Kuhn E, Kurman RJ, Sehdev AS et al. Ki-67 labeling index as an adjunct in the siagnosis of serous tubal Intraepithelial carcinoma. Int J Gynecol Pathol 2012;31:416–422.
Kuhn E, Kurman RJ, Vang R et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma-evidence supporting the clonal relationship of the two lesions. J Pathol 2012;226:421–426.
Kuhn E, Kurman RJ, Soslow RA et al. The diagnostic and biological implications of laminin expression in serous tubal intraepithelial carcinoma. Am J Surg Pathol 2012;36:1826–1834.
Kuhn E, Meeker AK, Visvanathan K et al. Telomere length in different histologic types of ovarian carcinoma with emphasis on clear cell carcinoma. Mod Pathol 2011;24:1139–1145.
Kuhn E, Kurman RJ, Shih IM . Ovarian cancer is an imported disease: fact or fiction? Curr Obstet Gynecol Rep 2012;1:1–9.
Shah SP, Kobel M, Senz J et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med 2009;360:2719–2729.
Cuatrecasas M, Catasus L, Palacios J et al. Transitional cell tumors of the ovary: a comparative clinicopathologic, immunohistochemical, and molecular genetic analysis of Brenner tumors and transitional cell carcinomas. Am J Surg Pathol 2009;33:556–567.
Ali RH, Seidman JD, Luk M et al. Transitional cell carcinoma of the ovary is related to high-grade serous carcinoma and is distinct from malignant brenner tumor. Int J Gynecol Pathol 2012;31:499–506.
Li J, Poi MJ, Tsai MD . Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011;50:5566–5582.
Hunter SM, Gorringe KL, Christie M et al. Pre-invasive ovarian mucinous tumors are characterized by CDKN2A and RAS pathway aberrations. Clin Cancer Res 2012;18:5267–5277.
Gorringe KL, Campbell IG . Large-scale genomic analysis of ovarian carcinomas. Mol Oncol 2009;3:157–164.
Seidman JD, Khedmati F . Exploring the histogenesis of ovarian mucinous and transitional cell (Brenner) neoplasms and their relationship with Walthard cell nests: a study of 120 tumors. Arch Pathol Lab Med 2008;132:1753–1760.
Kang S, Bader AG, Vogt PK . Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA 2005;102:802–807.
Forbes SA, Bindal N, Bamford S et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39, Database issue D945–D950.
Kurman RJ, Shih IeM . The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol 2010;34:433–443.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Kuhn, E., Ayhan, A., Shih, IM. et al. The pathogenesis of atypical proliferative Brenner tumor: an immunohistochemical and molecular genetic analysis. Mod Pathol 27, 231–237 (2014). https://doi.org/10.1038/modpathol.2013.142
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2013.142
Keywords
This article is cited by
-
Borderline Brenner tumor with abnormally high serum level of carbohydrate antigen 199: a rare case report and literature review
Irish Journal of Medical Science (1971 -) (2022)
-
Recurrent urothelial carcinoma-like FGFR3 genomic alterations in malignant Brenner tumors of the ovary
Modern Pathology (2021)
-
Ovarian borderline tumors in the 2014 WHO classification: evolving concepts and diagnostic criteria
Virchows Archiv (2017)
-
Borderline Brenner tumor of the ovary: a case report with immunohistochemical and molecular study
Journal of Ovarian Research (2014)
