
Abstract
Several recent reports have described low-grade fibromyxoid sarcoma with sclerosing epithelioid fibrosarcoma-like areas. We evaluated cases of pure sclerosing epithelioid fibrosarcoma lacking areas of low-grade fibromyxoid sarcoma for FUS rearrangement to determine whether this entity could be related to low-grade fibromyxoid sarcoma. Available formalin-fixed paraffin-embedded tissue of 27 sclerosing epithelioid fibrosarcoma from 25 patients was retrieved and tabulated with clinical information. Unstained slides from formalin-fixed paraffin-embedded blocks were prepared and fluorescence in-situ hybridization was performed using a commercial FUS break-apart probe. The median patient age at presentation was 50 (range, 14–78) years, with 14 males and 10 females. Sclerosing epithelioid fibrosarcoma most commonly involved the extremities (n=8) or chest (n=6). Sixteen patients had a median follow-up of 17 (range, 1–99) months; seven were alive and well at 12 (range, 5–30) months; three alive with disease at 28 (range, 9–99) months; five dead of disease at a median of 22 (range, 1–36) months and one was dead of unknown causes. Twelve patients were known to have metastases; the most common site was lung (n=7), followed by bone (n=3), lymph nodes (n=2) and peritoneum (n=1). Only 2 of 22 (9%) analyzable cases of sclerosing epithelioid fibrosarcoma showed rearrangement in the FUS locus by fluorescence in-situ hybridization. Although cytogenetically confirmed low-grade fibromyxoid sarcoma can have sclerosing epithelioid fibrosarcoma-like areas, FUS rearrangement, which is characteristic of low-grade fibromyxoid sarcoma, appears to be relatively rare in pure sclerosing epithelioid fibrosarcoma.
Similar content being viewed by others
Main
Sclerosing epithelioid fibrosarcoma is a rare sarcoma characterized by epithelioid cells arranged in nests and cords in a distinctly sclerotic hyalinized stroma that can mimic infiltrating carcinoma or sclerosing lymphoma. These tumors preferentially affect adults (median 45; range 14–87 years of age), and often involve the deep soft tissue of the lower extremities, although a wide age and site spectrum has been described. According to the two largest series published to date, approximately half of the patients developed recurrences with variable metastatic rates of 43 and 86%. The most common site of metastases was lung. Mortality rates of 25 and 57% have been reported.1, 2
Some investigators have suggested that there is a relationship between sclerosing epithelioid fibrosarcoma and low-grade fibromyxoid sarcoma. Low-grade fibromyxoid sarcoma is characterized by sweeping fibrous areas composed of relatively banal cells with myxoid zones. It harbors a recurrent translocation involving FUS (16p11), CREB3L2 (7q32–34), or CREB3L1 (7p11). Tumors previously described as ‘hyalinizing spindle cell tumor with giant rosettes’ are now recognized to be within the morphological spectrum of low-grade fibromyxoid sarcoma, as they have been found to harbor an identical reciprocal chromosomal translocation.3, 4, 5, 6, 7 Interestingly, in their study connecting both low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes as the same tumor, Reid et al8 discussed one case of hyalinizing spindle cell tumor with giant rosettes that had an sclerosing epithelioid fibrosarcoma-like area.
Early studies have noted areas reminiscent of sclerosing epithelioid fibrosarcoma in some cases of low-grade fibromyxoid sarcoma and vice versa.1, 2, 3, 4, 6, 7, 9, 10, 11 In the initial study of sclerosing epithelioid fibrosarcoma by Meis and Enzinger, rare spindled areas resembling various fibrous tumors, including low-grade fibromxyoid sarcoma, were mentioned.1 This observation was corroborated by Antonescu et al2 in their subsequent series. Large studies have occasionally reported sclerosing epithelioid fibrosarcoma-like areas in low-grade fibromyxoid sarcoma.7, 9, 10, 11 Folpe et al7 have described a case of hyalinizing spindle cell tumor with giant rosettes in which the metastasis had features reminiscent of sclerosing epithelioid fibrosarcoma, and noted cases of low-grade fibromyxoid sarcoma that had more epithelioid morphological features, as have members of the French Sarcoma Group.9, 10, 12 Evans10 also described one case of low-grade fibromyxoid sarcoma that had a recurrence with sclerosing epithelioid fibrosarcoma-like features, whereas Rekhi et al11 described sclerosing epithelioid fibrosarcoma-like areas in 6 of their 18 cases of low-grade fibromyxoid sarcoma.
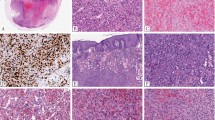
Some of the aforementioned studies have reported sclerosing epithelioid fibrosarcoma harboring the same translocation as low-grade fibromyxoid sarcoma.9, 12 The French Sarcoma Group found that four of their seven cases with sclerosing epithelioid fibrosarcoma features had the same chimeric fusion transcript as low-grade fibromyxoid sarcoma by RT-PCR in their study characterizing low-grade fibromyxoid sarcoma and non-low-grade fibromyxoid sarcoma tumors for FUS/CREB3L1/2.9 FUS rearrangements by fluorescence in-situ hybridization have also been described in a few other sclerosing epithelioid fibrosarcomas.12, 13 We have also observed cases of low-grade fibromyxoid sarcoma having sclerosing epithelioid fibrosarcoma-like areas, with both conventional low-grade fibromyxoid sarcoma and sclerosing epithelioid fibrosarcoma-like areas harboring rearrangement of FUS (16p11) by fluorescence in-situ hybridization (Figure 1).

Example of low-grade fibromyxoid sarcoma with sclerosing epithelioid-like areas (not included in the study). (a and b) Alternating zones of fibrous and myxoid areas with banal spindled cells are characteristic of low-grade fibromyxoid sarcoma. (c and d) More sclerotic hyalinized areas with epithelioid cells arranged in cords reminiscent of sclerosing epithelioid fibrosarcoma were also encountered. Both areas were positive for rearrangement in FUS.
Given that low-grade fibromyxoid sarcoma can include morphological variants such as hyalinizing spindle cell tumor with giant rosettes, we examined cases of pure sclerosing epithelioid fibrosarcoma without identifiable areas of low-grade fibromyxoid sarcoma for the presence of FUS rearrangement to determine whether sclerosing epithelioid fibrosarcoma was also a part of the morphological spectrum of low-grade fibromyxoid sarcoma.
Materials and methods
With institutional review board approval, 27 specimens of sclerosing epithelioid fibrosarcoma from 25 patients (two specimens were from the same patients), with formalin-fixed paraffin-embedded tissue available were retrieved and reviewed from the pathology files of The University of Texas MD Anderson Cancer Center, Cleveland Clinic, Medical University of Graz and Leiden University Medical Center from 1986 to 2010. Clinical information was obtained whenever available. These consisted of well-sampled cases where potential mimics had been excluded by immunohistochemical or molecular means as appropriate, and where the characteristic features of epithelioid cells within a fibrotic stromal was evident throughout sampled sections.
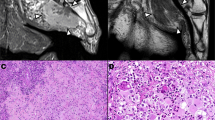
Fluorescence in-situ hybridization for 16q11 to detect FUS rearrangement was performed using the LSI FUS dual-color, break-apart probe (Abbott Molecular/Vysis, Des Plaines, IL, USA) according to the manufacturer's recommendations (Figure 2). Tissue sections (4-μm thick) were placed onto slides, air-dried, and baked overnight at 60°C. Slides were de-paraffinized in CitriSolv (Fisher, Vernon Hills, IL, USA) three times for 5 min, and then immersed in 100% ethanol twice for 1 min. After air-drying, slides were treated in Paraffin Pretreatment solution (Paraffin Pretreatment Kit II; Abbott Molecular/Vysis) for 10 min at 80°C, washed with purified water for 3 min at room temperature, and treated with protease solution for 15 min, at 37°C. Slides were subsequently rinsed in purified water for 3 min, air-dried, and put in 2 × saline-sodium citrate buffer at 37°C for 30 min, dehydrated in 70, 85, and 100% ethanol, respectively, and allowed to air-dry. Next, 10–20 μl of LSI FUS dual-color break-apart probe (Abbott Molecular/Vysis, Des Plaines, IL, USA) was applied to the slides in an approximately 1 cm2 area selected for a pure tumor population (>90% tumor cells), and hybridization was performed at 37°C overnight in a moist chamber. Excess probe was washed away using 2 × saline-sodium citrate buffer/0.3% NP-40 (Fisher) at 73°C for 2 min, and the nuclei were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride/Vectashield (Vector Laboratories, Burlingame, CA, USA).

Schematic and example of fluorescence in-situ hybridization on sclerosing epithelioid fibrosarcoma using FUS break-apart probe set. Two fluorescently labeled probes (one (R)ed and one (G)reen) hybridize to regions of DNA flanking the FUS loci. (a) In cells negative for FUS rearrangement, green and red signals overlap creating a yellow signal. Case 16 (inset) was negative for FUS rearrangement. (b) Cases positive for FUS rearrangement have cells with separated green and red signals (split signals) and one yellow (fused) signal resulting from of the spectral overlap of the green and red probes. Case 20 (inset) was positive for FUS rearrangement.
Hybridized tissue sections were examined under a Zeiss fluorescence microscope (Carl Zeiss, Thornwood, NY, USA) using a × 10 ocular, and × 63 and × 100 oil immersion lenses, and 100 nuclei evaluated for the presence of fused (yellow) or split (green and red) signals. Nuclei with one green, and one red (split) and one yellow (fused) signals detected simultaneously were considered positive for FUS rearrangement. Signals were considered to be colocalized when their distance was equal to or smaller than twice the diameter of the hybridization signal.
Samples were evaluated for the presence of fused or split signals in tumor cells, and an estimated percentage reported. A positive result was reported when >20% of tumor cells showed evidence of a rearrangement (split signal). The relatively high cut-off (we usually use 5% as a cut-off) was set to allow more rigorous examination of cases falling within the 5–20% range. However, this turned out to be unnecessary, as all of the positive cases in this series showed >50% rearrangement. When testing was not interpretable in a particular sample, it was repeated with increased proteinase digestion.
Results
Clinical information was available in 24 of 25 patients, whose tumors were studied (Table 1). The male to female ratio was 14:10. The median age was 50 years (range, 14–78). The most common involved sites were extremities (n=8) and chest (n=6), with other sites including retroperitoneum/abdomen (n=4), back (n=4), and head and neck (n=2). Median follow-up in 16 patients was 17 months (range, 1–99 months). Seven (43%) were alive and well at a median of 12 months (range, 9–30 months), three alive with disease at a median of 28 (range, 9–99 months), five dead of disease (31%) at a median of 22 months (range, 0.6–36 months), and one was dead of unknown causes at 51 months. Twelve patients were known to have metastases with the most common site being lung (n=7), followed by bone (n=3), lymph node (n=2), and peritoneum (n=1).
Twenty-four of 27 tested tumors, including two specimens from the same patient, were analyzed by fluorescence in-situ hybridization, with three failing despite repeat hybridization and additional proteinase digestion. Tumors from 22 patients were analyzable by fluorescence in-situ hybridization; of these, two patients (9%) had tumors positive for rearrangement of FUS. Both tumors lacked recognizable low-grade fibromyxoid sarcoma areas despite extensive sampling (Table 1; histological examples of cases shown in Figures 3 and 4).

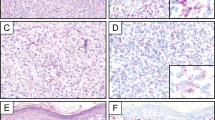
Case 16. (a–c) Sections reveal a well-circumscribed tumor composed of epithelioid cells, with angulated nuclei and arranged in cords with dense sclerotic stroma. This case was negative for FUS rearrangement.

Case 20. (a and b) Histological features of sclerosing epithelioid fibrosarcoma were seen without low-grade fibromyxoid sarcoma-like areas. This case was positive for FUS rearrangement.
Conclusion
This is the largest molecular study analyzing the presence of FUS rearrangement in tumors with well-sampled, pure sclerosing epithelioid fibrosarcoma histology to date. The total number of previously reported cases does not exceed 10. Rearrangement by fluorescence in-situ hybridization was seen in only 2 of 22 (9%) patients with sclerosing epithelioid fibrosarcoma in our series. In comparison, Guillou et al9 found that 4/7 (57%) of their sclerosing epithelioid fibrosarcomas harbored the characteristic translocation of low-grade fibromyxoid sarcoma by RT-PCR. Examination of FUS rearrangement by fluorescence in-situ hybridization in sclerosing epithelioid fibrosarcomas was less successful in their series; however, other studies have detected FUS rearrangement by fluorescence in-situ hybridization in sclerosing epithelioid fibrosarcoma.12 Some of the discrepancies in the percentage of cases harboring FUS rearrangements may be due, in part, to the relatively small number of cases reported in other series. Our study lacked adequate material for RT-PCR testing and many of the specimens were >5 years, with a high likelihood of degraded RNA. As previously noted, sclerosing epithelioid fibrosarcoma can have areas reminiscent of low-grade fibromyxoid sarcoma. However, in our series, cases with obvious low-grade fibromyxoid sarcoma-like areas were specifically excluded. These findings suggest that pure sclerosing epithelioid fibrosarcomas (tumors that lack recognizable low-grade fibromyxoid sarcoma-like areas) do not usually harbor FUS gene rearrangement, and could also explain the discrepancies in the percentage of FUS-positive cases between our series and other reports. The results also raise the possibility that tumors with focal sclerosing epithelioid fibrosarcoma-like areas in low-grade fibromyxoid sarcoma are best viewed—as within the spectrum of low-grade fibromyxoid sarcoma, though the natural history of such combined histology tumors is uncertain and in need of further study.
Other studies, albeit only a few, that have characterized the genetic alterations in sclerosing epithelioid fibrosarcoma have also failed to demonstrate rearrangements in t(7;16), seen in low-grade fibromyxoid sarcoma.14, 15, 16, 17 Cytogenetic analysis in one case of sclerosing epithelioid fibrosarcoma was described to harbor a 12q13–15 amplification, whereas another demonstrated a potential amplification of MDM2 by immunohistochemical studies.14, 17 Rearrangements in 10p11 have been reported in the cytogenetic analysis of two cases14, 16 and interestingly, a case reported to be low-grade fibromyxoid sarcoma was also found to have a rearrangement involving t(10;17)(p11;q23).18 No t(7;16) translocation was described in that case; whether this is an alternative translocation associated with low-grade fibromyxoid sarcoma is not clear. FUS rearrangements can be detected in up to 90%19, 20, 21 of low-grade fibromyxoid sarcoma by fluorescence in-situ hybridization, whereas FUS/CREB3L1/2 fusion transcripts have been reported to be detected in up to 96% of low-grade fibromyxoid sarcoma by RT-PCR.9, 21, 22, 23 Therefore, there may be other genetic aberration/s that could conceivably link a subset of sclerosing epithelioid fibrosarcoma to low-grade fibromyxoid sarcoma. However, the prevalence of t(7;16) is substantially lower in sclerosing epithelioid fibrosarcoma than in low-grade fibromyxoid sarcoma in this present study.
It is also possible that some sclerosing epithelioid fibrosarcomas may share the same recurrent translocation as low-grade fibromyxoid sarcomas, but still be unrelated pathogenetically. Other rearrangements involving identical loci and the same recurrent translocations can be shared by different sarcomas, lymphomas, and carcinomas.24, 25, 26 Similar to EWSR1, rearrangements in FUS have been reported in multiple sarcomas, including low-grade fibromyxoid sarcoma, myxoid liposarcoma, angiomatoid fibrous histiocytomas, and Ewing sarcoma.25 Both angiomatoid fibrous histiocytoma and clear cell sarcoma harbor the same two translocation variants, EWSR1-ATF1 t(12;22)(q13;q12) and EWSR1-CREB1 t(2;22)(q34;q12), yet both are phenotypically distinct from each other with distinctly different clinical courses.27, 28, 29, 30 More recently, recurrent EWSR1–CREB1 translocations have also been identified in hyalinizing clear cell carcinoma of the salivary gland.26 Other examples include involvement of ALK–TPM3 in inflammatory myofibroblastic tumor and anaplastic large cell lymphomas, and ETV6–NTRK in infantile fibrosarcoma, secretory breast carcinomas, and acute myeloid leukemia.24, 31 These findings challenge the previously held notion that recurrent translocations are unique to a particular sarcoma or tumor class. A similar scenario is possible for sclerosing epithelioid fibrosarcoma and low-grade fibromyxoid sarcoma. However, the presence of prominent sclerosing epithelioid fibrosarcoma-like areas in some low-grade fibromyxoid sarcoma, including metastases, seems to suggest that there is a small subset of tumors that appear to be sclerosing epithelioid fibrosarcoma, but are really low-grade fibromyxoid sarcomas lacking recognizable conventional areas owing to limited sampling, or perhaps overgrowth of the sclerosing epithelioid fibrosarcoma-like areas. Other sarcomas can have areas that mimic sclerosing epithelioid fibrosarcoma including osteosarcoma and synovial sarcoma among others, but generally will have other more conventional areas. Carcinomas and even melanoma can sometimes also be associated with extensive sclerosis, but immunohistochemistry can readily exclude these potential mimics.
In our series, 5 of 16 patients (31%) with available clinical follow-up died of disease within a median time span of 2 years, and approximately half of our patients were known to develop metastases. Although the interval to metastasis is shorter, the mortality and metastatic rates are similar to those previously reported (43–86 and 25–57%, respectively) from the two largest sclerosing epithelioid fibrosarcoma series.1, 2 Variability in rates may be partially explained by the limitations of small studies of a rare tumor as well as patient referral population or intrinsic bias (ie, major sarcoma treatment center versus consultative practice). In comparison, large series on low-grade fibromyxoid sarcoma have reported mortality rates of 2–40% and metastatic rates of 6–45%, with higher rates correlating with longer follow-up.7, 9, 10 These wide ranges are likely due to the variability in follow-up between studies, as patients with low-grade fibromyxoid sarcoma are known to develop metastasis after several years. Evans10 reported a median of 5 years for metastasis, and Guillou et al9 state that the majority of their patients developed metastases after 9 years. Evans10 also found that patients died of disease after a prolonged period of time (median 11 years) and that those developing dedifferentiation had a much shorter survival after tumor progression.
The clinical significance of sclerosing epithelioid fibrosarcoma-like areas in low-grade fibromyxoid sarcoma is not certain. Rekhi et al11 reported that four of their six patients with sclerosing epithelioid fibrosarcoma-like areas were alive, with one developing a recurrence. Reid et al8 also describe one case of a hyalinizing spindle cell tumor with giant rosettes, with sclerosing epithelioid fibrosarcoma-like features; this patient was alive and well. However, follow-up in these studies was relatively short (5–52 months).11 Sclerosing epithelioid fibrosarcoma-like areas may be a spectrum of the process seen in the giant rosettes. In our series, only one of the two patients with sclerosing epithelioid fibrosarcoma positive for FUS rearrangement had follow-up. That patient was alive but had a lung metastasis 9 months after the primary diagnosis. Additional studies with long-term follow-up are needed to compare the impact of sclerosing epithelioid fibrosarcoma-like areas in low-grade fibromyxoid sarcoma or pure sclerosing epithelioid fibrosarcoma with FUS rearrangement to low-grade fibromyxoid sarcoma lacking this feature.
In summary, we describe a series of sclerosing epithelioid fibrosarcomas that lacked recognizable areas of low-grade fibromyxoid sarcoma. These cases lacked FUS rearrangement by fluorescence in-situ hybridization for the most part. The presence of FUS rearrangements in a minority of sclerosing epithelioid fibrosarcomas and the presence of tumors with hybrid areas suggest that a subset of cases of sclerosing epithelioid fibrosarcomas may represent low-grade fibromyxoid sarcoma, with a prominent sclerosing epithelioid fibrosarcoma-like area (perhaps with low-grade fibromyxoid sarcoma areas not sampled and/or focal). However, the majority of sclerosing epithelioid fibrosarcoma tested in our study do not appear to be genetically related to low-grade fibromyxoid sarcoma as assessed by FUS gene rearrangements.
References
Meis-Kindblom JM, Kindblom LG, Enzinger FM . Sclerosing epithelioid fibrosarcoma. A variant of fibrosarcoma simulating carcinoma. Am J Surg Pathol 1995;19:979–993.
Antonescu CR, Rosenblum MK, Pereira P, et al. Sclerosing epithelioid fibrosarcoma: a study of 16 cases and confirmation of a clinicopathologically distinct tumor. Am J Surg Pathol 2001;25:699–709.
Meis-Kindblom JM, Kindblom LG, van den Berg E, et al. Sclerosing epithelioid fibrosarcoma. In: Fletcher CDM, Unni KK, Mertens FF (eds). Pathology and Genetics of Tumours of Soft Tissue and Bone. IARCPress: Lyon France, 2002, pp 106–107.
Weiss SR, Goldblum JR, (eds). Enzinger and Weiss's Soft Tissue Tumors, 5th edn. Mosby Elsevier: St Louis, 2008, pp 310–325.
Lane KL, Shannon RJ, Weiss SW . Hyalinizing spindle cell tumor with giant rosettes: a distinctive tumor closely resembling low-grade fibromyxoid sarcoma. Am J Surg Pathol 1997;21:1481–1488.
Woodruff JM, Antonescu CR, Erlandson RA, et al. Low-grade fibrosarcoma with palisaded granulomalike bodies (giant rosettes): report of a case that metastasized. Am J Surg Pathol 1999;23:1423–1428.
Folpe AL, Lane KL, Paull G, et al. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol 2000;24:1353–1360.
Reid R, de Silva MV, Paterson L, et al. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes share a common t(7;16)(q34;p11) translocation. Am J Surg Pathol 2003;27:1229–1236.
Guillou L, Benhattar J, Gengler C, et al. Translocation-positive low-grade fibromyxoid sarcoma: clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma: a study from the French Sarcoma Group. Am J Surg Pathol 2007;31:1387–1402.
Evans HL . Low-grade fibromxyoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 2011;35:1450–1462.
Rekhi B, Deshmukh M, Jambhekar NA . Low-grade fibromyxoid sarcoma: a clinicopathologic study of 18 cases, including histopathologic relationship with sclerosing epithelioid fibrosarcoma in a subset of cases. Ann Diagn Pathol 2011;15:303–311.
Rekhi B, Folpe AL, Deshmukh M, et al. Sclerosing epithelioid fibrosarcoma—a report of two cases with cytogenetic analysis of FUS gene rearrangement by FISH technique. Pathol Oncol Res 2011;17:145–148.
Elkins CT, Wakely Jr PE . Sclerosing epithelioid fibrosarcoma of the oral cavity. Head Neck Pathol 2011;5:428–423.
Gisselsson D, Andreasson P, Meis-Kindblom JM, et al. Amplification of 12q13 and 12q15 sequences in a sclerosing epithelioid fibrosarcoma. Cancer Genet Cytogenet 1998;107:102–106.
Donner LR, Clawson K, Dobin SM . Sclerosing epithelioid fibrosarcoma: a cytogenetic, immunohistochemical, and ultrastructural study of an unusual histological variant. Cancer Genet Cytogenet 2000;119:127–131.
Ogose A, Kawashima H, Umezu H, et al. Sclerosing epithelioid fibrosarcoma with der(10)t(10;17)(p11;q11). Cancer Genet Cytogenet 2004;152:136–140.
Jiao YF, Nakamura S, Sugai T, et al. Overexpression of MDM2 in a sclerosing epithelioid fibrosarcoma: genetic, immunohistochemical and ultrastructural study of a case. Pathol Int 2002;52:135–140.
Sawyer JR, Binz RL, Gilliland JC, et al. A novel reciprocal (10;17)(p11.2;q23) in myxoid fibrosarcoma. Cancer Genet Cytogenet 2001;124:144–146.
Downs-Kelly E, Goldblum JR, Patel RM, et al. The utility of fluorescence in situ hybridization (FISH) in the diagnosis of myxoid soft tissue neoplasms. Am J Surg Pathol 2008;32:8–13.
Patel RM, Downs-Kelly E, Dandekar MN, et al. FUS (16p11) gene rearrangement as detected by fluorescence in-situ hybridization in cutaneous low-grade fibromyxoid sarcoma: a potential diagnostic tool. Am J Dermatopathol 2011;33:140–143.
Weindel M, Wang WL, Demicco EG, et al. Genetic characterization and MUC4 expression in a series of histiologically characteristic low-grade fibromxyoid sarcoma [Abstract]. Mod Pathol 2011;24:23A.
Matsuyama A, Hisaoka M, Shimajiri S, et al. Molecular detection of FUS-CREB3L2 fusion transcripts in low-grade fibromyxoid sarcoma using formalin-fixed, paraffin-embedded tissue specimens. Am J Surg Pathol 2006;30:1077–1084.
Mertens F, Fletcher CD, Antonescu CR, et al. Clinicopathologic and molecular genetic characterization of low-grade fibromyxoid sarcoma, and cloning of a novel FUS/CREB3L1 fusion gene. Lab Invest 2005;85:408–415.
Miettinen M . From morphological to molecular diagnosis of soft tissue tumors. Adv Exp Med Biol 2006;587:99–113.
Riggi N, Cironi L, Suva ML, et al. Sarcomas: genetics, signalling, and cellular origins. Part 1: the fellowship of TET. J Pathol 2007;213:4–20.
Antonescu CR, Katabi N, Zhang L, et al. EWSR1-ATF1 fusion is a novel and consistent finding in hyalinizing clear-cell carcinoma of salivary gland. Genes Chromosomes Cancer 2011;50:559–570.
Antonescu CR, Dal Cin P, Nafa K, et al. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 2007;46:1051–1060.
Antonescu CR, Nafa K, Segal NH, et al. EWS-CREB1: a recurrent variant fusion in clear cell sarcoma—association with gastrointestinal location and absence of melanocytic differentiation. Clin Cancer Res 2006;12:5356–5362.
Antonescu CR, Tschernyavsky SJ, Woodruff JM, et al. Molecular diagnosis of clear cell sarcoma: detection of EWS-ATF1 and MITF-M transcripts and histopathological and ultrastructural analysis of 12 cases. J Mol Diagn 2002;4:44–52.
Rossi S, Szuhai K, Ijszenga M, et al. EWSR1-CREB1 and EWSR1-ATF1 fusion genes in angiomatoid fibrous histiocytoma. Clin Cancer Res 2007;13:7322–7328.
Kralik JM, Kranewitter W, Boesmueller H, et al. Characterization of a newly identified ETV6-NTRK3 fusion transcript in acute myeloid leukemia. Diagn Pathol 2011;6:19.
Acknowledgements
We thank Kim Vu for expert assistance in graphic art.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Wang, WL., Evans, H., Meis, J. et al. FUS rearrangements are rare in ‘pure’ sclerosing epithelioid fibrosarcoma. Mod Pathol 25, 846–853 (2012). https://doi.org/10.1038/modpathol.2011.214
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.214
Keywords
This article is cited by
-
Sclerosing epithelioid fibrosarcoma of bone with hybrid features: clinicopathologic, radiologic, and molecular analysis of three cases
Skeletal Radiology (2024)
-
Sclerosing epithelioid fibrosarcoma of bone: morphological, immunophenotypical, and molecular findings of 9 cases
Virchows Archiv (2021)
-
What’s new in fibroblastic tumors?
Virchows Archiv (2020)
-
Primary pulmonary clear cell sarcoma—the first two reported cases
Virchows Archiv (2016)
