
Abstract
The tumor necrosis factor (TNF) superfamily protein TNF-like 1A (TL1A) is the ligand for death receptor 3 (DR3). TL1A is induced on activated dendritic cells (DCs) and its expression has been linked to human inflammatory bowel disease. To address how TL1A might influence intestinal inflammation, we generated transgenic mice that constitutively express TL1A on DCs. TL1A transgenic mice developed striking goblet cell hyperplasia in the ileum that was associated with elevated interleukin (IL)-13 levels in the small intestine. IL-13- and IL-17-producing small intestinal lamina propria T cells were increased in TL1A transgenic mice. TL1A also enhanced regulatory T (Treg) cell turnover in vivo and directly stimulated Treg cell proliferation in vitro. The presence of TL1A attenuated the ability of Treg cells to suppress conventional T cells, an effect that required DR3 signaling in either conventional T cells or Treg cells. Our findings identify mechanisms by which chronic DR3 signaling could promote pathogenesis in inflammatory bowel disease.
Similar content being viewed by others


Introduction
An inappropriate effector T-cell response to the commensal flora is considered to be an important feature in the pathogenesis of inflammatory bowel disease.1 Effector T-cell responses directed against the commensal microbiota are normally restrained by regulatory cells, therefore limiting the potential for an overt inflammatory response.2 In particular, CD4+Foxp3+ regulatory T (Treg) cells have a key role in promoting tolerance to the commensal flora and dietary antigens and are essential in maintaining tolerance to self-antigens.2, 3 Although most CD4+Foxp3+ Treg cells are thought to originate in the thymus, inducible Foxp3+ Treg cells can be generated from naive CD4+ T cells in certain microenvironments, including the gut-associated lymphoid tissue.2 Although hyporesponsive to T-cell receptor stimulation in vitro,4 Treg cells proliferate and expand in vivo following exposure to self or pathogen-derived antigens.5, 6, 7 In addition to T-cell receptor signaling, CD28 costimulation has been shown to be important for proliferation of self-reactive Treg cells, a conclusion supported by in vitro studies using antigen-loaded dendritic cells (DCs).8 There is also evidence that triggering of other costimulatory receptors, including GITR (glucocorticoid-induced TNF receptor),9 OX40,10 and 4-1BB,11 can promote Treg cell expansion, but it remains to be determined if this effect is because of direct stimulation of Treg cells or is mediated indirectly, for example, by interleukin (IL)-2 produced by activated conventional T cells. In addition, stimulation via OX40 on Treg cells has been reported to block their suppressive function,12 whereas GITR signaling in conventional T cells can render them resistant to suppression by Treg cells.13
Death receptor 3 (DR3) is a member of the tumor necrosis factor (TNF) receptor superfamily that shows the highest homology with TNF receptor-1. Unlike TNF receptor-1, which is ubiquitously expressed, DR3 is expressed primarily on T cells. DR3 is expressed constitutively on CD4+ T cells, including Treg cells, and is upregulated by antigen stimulation.14, 15 A stimulatory role for DR3 in CD4+ effector T-cell responses has been suggested based on in vitro16, 17, 18, 19 and more recently in vivo studies.14, 15, 19 Thus, in mice that are deficient in DR3 signaling, accumulation of effector CD4+ T cells within the inflamed target tissue was reduced, and consequently immunopathology associated with experimental autoimmune encephalomyelitis and allergic lung inflammation was ameliorated.14, 15, 19 The TNF-like protein TL1A (TNFSF15) is the ligand for DR3.16 TL1A is expressed following activation of DCs and monocytes by immune complexes20 or Toll-like receptor (TLR) ligands.19 In addition, TL1A is upregulated on T-cell receptor-stimulated T cells and on endothelial cells exposed to IL-1 or TNF-α.14, 16, 19
TL1A expression is tightly regulated following TLR stimulation in vitro19 and in vivo (this study). However, sustained expression of TL1A was detected on lamina propria DCs in two spontaneous mouse models of chronic ileitis,18 and both TL1A and DR3 are upregulated in human inflammatory bowel disease.21 To address how sustained expression of TL1A could influence immune responses and inflammation in vivo, we generated transgenic mice that express TL1A constitutively under the control of the CD11c promoter (CD11c-TL1A tg mice). These mice developed striking goblet cell hyperplasia in the ileum that was associated with increased production of IL-13 in the intestinal tissue and an increase in the frequency of activated/memory CD4+ effector T cells. Surprisingly, TL1A exerted stimulatory effects on Treg cell turnover, but overall the presence of TL1A attenuated the ability of Treg cells to suppress conventional T cells. These data reveal novel roles for TL1A in regulating T cell-mediated immune responses that have implications in the pathogenesis of inflammatory bowel disease.
Results
Sustained expression of TL1A in CD11c-TL1A tg mice
Previous work has shown that in vitro activation of DCs by the TLR ligands lipopolysaccharide or soluble tachyzoite antigen from Toxoplasma gondii upregulates TL1A mRNA.19 To examine the effect of DC activation on the expression of TL1A in vivo, we injected C57BL/6 mice with various TLR ligands, the dectin-1 agonist curdlan, or agents that activate DCs via CD40 either directly (anti-CD40 antibody) or indirectly (α-galactosylceramide). Injection of lipopolysaccharide (TLR4 agonist) or polyinosinic–polycytidylic acid (TLR3 agonist) induced upregulation of TL1A mRNA with expression peaking at 4 and 6 h, respectively, and declining rapidly to background levels by 24 h (Figure 1a). In contrast, injection of unmethylated oligonucleotide CpG (TLR9 agonist), Pam3CysSerLys4 (TLR2 agonist), agonist anti-CD40 monoclonal antibody (mAb), α-galactosylceramide, or curdlan did not induce upregulation of TL1A (Figure 1a and not shown). Thus, TL1A expression in vivo is transient and is induced by a subset of receptors associated with DC activation. Expression of TL1A was then assessed in CD11c-TL1A tg mice. In naive transgenic mice, expression of TL1A mRNA in the spleen was approximately tenfold higher than that detected in naive control littermates (Figure 1a). Thus, the magnitude of TL1A mRNA expression in CD11c-TL1A tg mice was comparable to that seen at the peak of the lipopolysaccharide stimulation response in wild-type mice. TL1A mRNA was also present in the mesenteric lymph nodes and ilea of CD11c-TL1A tg mice, although at a lower level than that found in the spleen. Expression of TL1A protein in spleens from CD11c-TL1A tg mice was detected in scattered cells in the red pulp, in clusters of cells in and around the marginal zone, and in a few cells in the T-zone, consistent with the distribution of DCs in this organ (Figure 1b). A higher magnification image showed colocalization of TL1A with CD11c staining, confirming that the TL1A transgene is expressed in DCs. In naive non-transgenic mice, expression of TL1A was less intense and confined to a few cells in the red pulp and T-zone (Figure 1b).

Kinetics of TL1A expression following Toll-like receptor 3/4 (TLR3/4) triggering in vivo and expression levels in CD11c-TL1A transgenic (tg) mice. (a) Expression of TL1A transcripts in the spleens of 8–12-week-old tg CD11c-TL1A mice relative to that of wild-type (wt) mice as determined by quantitative reverse transcriptase-PCR (RT-PCR; solid bar) and kinetics of expression in wt mice after injection of lipopolysaccharide (LPS; 40 μg), polyinosinic–polycytidylic acid (pIC; 50 μg), or anti-CD40 monoclonal antibody (mAb; 500 μg). TL1A expression was not upregulated by administration of anti-CD40 mAb (data points represent the mean fold change of 3 mice per treatment per time point ± s.e. Fold change was calculated from triplicate quantitative PCR samples). (b) TL1A protein expression in wt and tg mice. Frozen wt and tg spleen sections were labeled with anti-mouse TL1A (red) and counterstained with anti-CD3 (blue) and anti-CD11c (green). The lower panel is a higher magnification image of TL1A-expressing cells in the red pulp and at the edge of the T-zone. TL1A expression colocalizes with CD11c staining (lower panel; orange). Bars=200 μm (top panels) and 50 μm (lower panel).
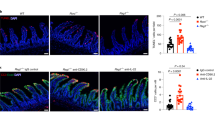
CD11c-TL1A tg mice were fertile, born at expected Mendelian frequencies, and appeared healthy. Because TL1A expression has been shown to be upregulated in Crohn's disease and ulcerative colitis21 and in mouse models of intestinal inflammation,18 we assessed whether CD11c-TL1A tg mice develop spontaneous intestinal inflammation. Histological examination of colons from CD11c-TL1A tg mice revealed normal tissue architecture and lack of inflammation (Figure 2a). Thus, there were no signs of epithelial cell hyperplasia, increased leukocyte infiltration, goblet cell depletion, or tissue ulceration in the colons of CD11c-TL1A tg mice. In contrast with the normal appearance of the colon, histological examination of the small intestine revealed a marked increase (two- to three fold) in the number of goblet cells in the ilea of CD11c-TL1A tg mice (Figure 2b). Further examination of sections from the small intestine revealed that there was also an increase in the number of Paneth cells in the ilea of the transgenic mice (Figure 2c). Thus, constitutive expression of TL1A promotes goblet cell and Paneth cell hyperplasia in the small intestine but does not lead to overt intestinal inflammation.

Sustained expression of TL1A leads to goblet cell and Paneth cell hyperplasia in the small intestine. Representative (n=10) photomicrographs (original magnification × 100) of sections from the colon (a) and ileum (b) of a 24-week-old CD11c-TL1A transgenic (tg) or wild-type (wt) mouse. (c) High-magnification ( × 400) photomicrographs of granule-rich Paneth cells from the ileum of a CD11c-TL1A tg or wt mouse. Paraffin sections stained with hematoxylin and eosin (H&E). The numbers of goblet cells/villus and Paneth cells/crypt were determined by examining 60 individual villi and crypts from 6 CD11c-TL1A tg or wt mice.
Enhanced T-cell activation in CD11c-TL1A tg mice
The total cellularity of spleens and lymph nodes from CD11c-TL1A tg mice was similar to that found in littermate control mice (not shown), and CD11c-TL1A tg mice had normal frequencies of CD4+ and CD8+ T cells, B cells, macrophages, DCs, and natural killer (NK) cells (Figure 3a). However, there was approximately a 50% decrease in the frequency of invariant natural killer T (NKT) cells in CD11c-TL1A tg mice (Figure 3a). To assess the effect of sustained TL1A expression on T-cell activation, we compared the expression of activation/memory T-cell markers on CD4+ T cells from transgenic and littermate controls. For these studies we electronically gated out Treg cells based on expression of Foxp3 (forkhead box P3) in order to examine the effect of TL1A specifically on conventional T cells. The percentage of CD69+, CD44high, and CD62Llow CD4+ T cells was significantly higher in transgenic mice when compared with control mice (Figure 3b), demonstrating that sustained expression of TL1A results in enhanced activation of conventional CD4+ T cells that presumably recognize environmental antigens.

Frequencies of leukocyte subsets in CD11c-TL1A transgenic (tg) mice and activation status of CD4+ T cells. (a) Representative (n=5) flow cytometric plots of spleen cells from tg or wild-type (wt) mice demonstrating similar frequencies of CD4+ T cells, CD8+ T cells, macrophages (CD11b+), dendritic cells (CD11c high), B cells (CD19+), and natural killer (NK) cells (NK1.1+CD3−) in tg and wt mice. However, fewer invariant natural killer T (NKT) cells (NK1.1+ CD3+ or CD1d/α-galactosylceramide (αGalCer) tetramer+) were present in CD11c-TL1A tg mice. (b) Representative flow cytometric plots showing expression of activation/memory markers on peripheral CD4+Foxp3− cells from wt and tg mice (7–13-week-old mice; the data shown are representative of five mice per group).
Constitutive expression of TL1A leads to a marked induction of IL-13 in the small intestine
Goblet cell and Paneth cell hyperplasia are processes that are associated with elevated T helper type 2 (Th2) cytokine production.22, 23 To assess whether Th2 cytokines are elevated in CD11c-TL1A tg mice, we analyzed expression of IL-4 and IL-13 transcripts. Expression of IL-4 was not increased in CD11c-TL1A tg mice; however, we observed a marked elevation of IL-13 in both the mesenteric lymph nodes (∼30-fold) and ilea (∼6-fold) of CD11c-TL1A tg mice (Figure 4a, b). The finding that IL-13, but not IL-4, levels were elevated is not unexpected, as others have reported that when compared with IL-4, IL-13 production is higher and more prolonged during Th2-polarized immune responses.24, 25 Interestingly, no increase in IL-13 transcripts was detected in the colons of CD11c-TL1A tg mice (Figure 4b). We also assessed whether constitutive TL1A expression influences IL-17 and interferon-γ (IFN-γ) levels. A relatively modest (approximately five fold), but statistically significant increase in IL-17 was detected in the mesenteric lymph nodes of CD11c-TL1A tg mice (Figure 4a). There was also a trend toward higher IL-17 expression in the ilea of CD11c-TL1A tg mice, but this was not statistically significant (Figure 4b). Surprisingly, IFN-γ expression did not differ between CD11c-TL1A tg and wild-type mice (Figure 4a). To address the contribution of CD4+ lamina propria lymphocytes (LPLs) to the increased expression of IL-13 and IL-17 in CD11c-TL1A tg mice, we isolated LPLs from the small intestines of transgenic and wild-type mice and assessed their ability to produce cytokines following stimulation with anti-CD3 and anti-CD28. Intracellular staining of IL-13, IL-17, and IFN-γ showed relatively modest increases in IL-13- and IL-17-producing cells among CD4+ LPLs in the small intestines of CD11c-TL1A tg mice (Figure 4c). In contrast, there was no increase in the frequency of IFN-γ-producing CD4+ LPLs in CD11c-TL1A tg. These data suggest that activated CD4+ T cells could represent a source for IL-13 and IL-17 in the small intestine of CD11c-TL1A tg mice, but do not exclude other cellular source for these cytokines.

Increased spontaneous expression of interleukin (IL)-13 and IL-17 in CD11c-TL1A transgenic (tg) mice. (a) Relative mRNA expression of IL-13, IL-17, interferon-γ (IFN-γ), and IL-4 in mesenteric lymph node (mLN) cells of CD11c-TL1A tg and wild-type (wt) mice (7–24 weeks old) as determined by quantitative real-time PCR. (b) Relative mRNA expression of IL-13 and IL-17 in intestinal tissues of CD11c-TL1A tg and wt mice as determined by quantitative real-time PCR. Each data point represents the relative cytokine expression value of an individual mouse. Relative expression values were calculated from triplicate quantitative PCR samples. (c) Expression of IL-13, IL-17, and IFN-γ by re-stimulated CD4+ lamina propria lymphocytes (LPLs) isolated from the small intestine of CD11c-TL1A tg and wt mice. Each data point represents cytokine expression in cells isolated from an individual mouse.
TL1A promotes Treg cell activation and proliferation
In the course of examining the effect of sustained expression of TL1A on the activation state of CD4+ T cells, we noticed an increase in the proportion of CD4+CD25+ T cells in CD11c-TL1A tg mice (not shown). Because recently activated conventional T cells as well as Treg cells express CD25, we used expression of the Treg marker, Foxp3, to delineate the effect of TL1A on Treg cells. The percentage of CD4+Foxp3+ T cells in secondary lymphoid organs of CD11c-TL1A tg mice was 1.7-fold higher than that found in control littermates (Figure 5a). Furthermore, the frequency of Treg cells among small intestinal CD4+ LPLs was also significantly increased in CD11c-TL1A tg mice (Figure 5b). Treg cells with a naive phenotype (CD44low CD62Lhigh CD69−) have been shown to proliferate and acquire an activated/memory phenotype in the periphery.5 In CD11c-TL1A tg mice, there was a clear and reproducible increase in the proportion of splenic Treg cells that expressed the proliferation antigen Ki-67 (Figure 5c). An increase in the Ki-67 antigen in CD4+Foxp3− T cells was also detected (Figure 5c), consistent with the observed increase in activation of conventional CD4+ T cells in CD11c-TL1A tg mice. We also found that the frequency of splenic Foxp3+ Treg cells with an activated phenotype was increased in CD11c-TL1A tg mice (Figure 5c). Together, these data suggest that TL1A promotes the proliferation of Treg cells. To address whether TL1A stimulates Treg cell proliferation by acting directly on Treg cells, we examined the ability of soluble recombinant TL1A (Figure 6a) to promote the proliferation of purified Treg cells in vitro. Consistent with previous studies,4 CD25+ Treg cells did not proliferate in the presence of anti-CD3 mAb; however, Treg cells were stimulated to proliferate when both anti-CD3 and anti-CD28 mAbs were present in the culture media (Figure 6b). The addition of TL1A to cultures of anti-CD3-stimulated CD4+ CD25+ T cells induced their proliferation, and the magnitude of the TL1A-stimulated response exceeded that observed with anti-CD28 mAb (Figure 6b). Furthermore, TL1A synergized with anti-CD28 mAb in driving Treg cell proliferation (Figure 6b). The effect of TL1A was completely abolished when CD4+ CD25+ T cells were purified from DR3-deficient mice, demonstrating that TL1A mediated its effects through DR3 (Figure 6b). To exclude the possibility that the effects of TL1A could have been because of stimulation of a small population of contaminating CD25+ conventional T cells, we repeated these experiments using highly purified Treg cells from Foxp3–GFP (green fluorescent protein) knock-in mice.26 that were isolated by cell sorting based on expression of GFP (Figure 6c). The findings from these experiments confirmed that TL1A triggers Treg cell proliferation by acting directly on Treg cells (Figure 6d).

Increased numbers and enhanced activation of regulatory T (Treg) cells in CD11c-TL1A transgenic (tg) mice. Bar graph representation of the frequency of Foxp3+ Treg cells in the (a) spleen and (b) small intestine of CD11c-TL1A tg and wild-type (wt) littermate control mice (6–13 weeks old; n=6–8 mice per group). (c) Increased turnover of Treg cells in CD11c-TL1A tg mice as defined by the frequency of proliferating (Ki-67+) peripheral CD4+Foxp3+ cells in tg and wt mice (8 weeks old; n=3 mice per group). (d) Flow cytometric plots of activation/memory markers on electronically gated splenic CD4+Foxp3+ cells from 8-week-old tg vs. wt littermate mice. Data are representative of six independent experiments using 6–13-week-old mice.

TL1A directly costimulates regulatory T (Treg) cell proliferation. (a) Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of purified soluble recombinant TL1A (sTL1A) confirms the predicted molecular mass of the recombinant fusion protein. (b) Wild-type (wt) or death receptor 3 (DR3)-deficient (DR3 KO) magnetically isolated CD4+CD25+ T cells (purity ≥95%) were stimulated in the presence of irradiated DR3 KO antigen-presenting cells (APCs) with anti-CD3 or anti-CD3/CD28. Where indicated, sTL1A was added and proliferation was determined at 72 h by 3H-thymidine (3H-TdR) incorporation. (c, d) sTL1A costimulates Foxp3+ Treg cells. (c) Dot-plots of Foxp3+ Treg cells from Foxp3–GFP knock-in mice pre- and post-cell sorting (purity ≥99.5%). (d) Foxp3+ Treg cells were stimulated in the presence of irradiated DR3 KO APCs with anti-CD3 or anti-CD3/CD28 and where indicated sTL1A was added. Bars represent the mean±s.e. of triplicate cultures. The results are representative of two independent experiments.
TL1A attenuates suppression by Treg cells
To address whether TL1A costimulation affects the ability of Treg cells to suppress conventional effector T cells, we co-cultured Treg cells and carboxyfluorescein succinimidyl ester-labeled conventional T cells in the presence of anti-CD3 with or without soluble recombinant TL1A. Addition of TL1A allowed conventional T cells to proliferate despite the presence of Treg cells (Figure 7). This showed that TL1A attenuates suppression by Treg cells. To address if blockade of suppression by TL1A was the result of DR3 signaling in conventional effector T cells or Treg cells, we used DR3-deficient and wild-type conventional or Treg cells in various combinations. Triggering of DR3 on conventional T cells alone was sufficient to overcome suppression by Treg cells, although less efficiently than when both conventional T cells and Treg cells expressed DR3 (Figure 7). When expression of DR3 was restricted to Treg cells, TL1A was also capable of attenuating the suppressive function of Treg cells (Figure 7). Taken together, these results show that triggering of DR3 in conventional effector CD4+ T cells and Treg cells contributes to the ability of TL1A to reverse the suppression exerted by Treg cells.

TL1A attenuates Treg cell-mediated suppression. Conventional effector CD4+CD25− T cells (effector T cells; Teff) and CD4+CD25+ T cells (regulatory T cells; Treg) were purified from wild-type (wt) or death receptor 3 (DR3) KO mice. Carboxyfluorescein succinimidyl ester (CFSE)-labeled wt or DR3 KO Teff were stimulated with anti-CD3 or anti-CD3 and soluble recombinant TL1A (sTL1A) in the absence or presence of titrated numbers of either wt or DR3 KO Treg. T-cell cultures also contained irradiated DR3 KO antigen-presenting cells (APCs). Teff proliferation at 72 h was analyzed by dilution of CFSE. The percentages of Teff cells that had divided are indicated in each plot. Data shown are representative of two independent experiments.
Discussion
A striking finding of this study is that constitutive expression of TL1A on DCs results in goblet cell and Paneth cell hyperplasia in the small intestine (Figure 2). Consistent with this finding, IL-13 was the most prominent cytokine that was elevated in the small intestinal tissue of CD11c-TL1A tg mice (Figure 4b). IL-13-induced goblet cell and Paneth cell hyperplasia constitutes a host-defense response that is normally elicited by gastrointestinal nematodes.22, 23, 27, 28, 29 Besides IL-13, there was a modest increase in IL-17 levels in the mesenteric lymph nodes of CD11c-TL1A tg mice, but no increase in IFN-γ expression (Figure 4). The pattern of cytokine expression and the lack of overt intestinal inflammation in CD11c-TL1A tg mice contrast with the phenotype of transgenic mice that constitutively express TNF-α or LIGHT, another TNF family protein, where elevated levels of IFN-γ were associated with severe intestinal immune pathology.30, 31 Furthermore, increased levels of IFN-γ were detected in mice that constitutively express CD70 or GITR ligand, two other TNF family proteins.9, 32 Interestingly, mice that constitutively express OX40 ligand also have a bias toward overproduction of Th2 cytokines, similar to what we observed in CD11c-TL1A tg mice, but additionally OX40 ligand transgenic mice spontaneously develop inflammation in the colon.33 Although currently unclear, the differences between the effects of TL1A and other TNF family proteins could be because of differences in the cellular distribution and signaling capacity of the respective receptors. It is noteworthy that transgenic mice that constitutively express TL1A on T cells also have increased levels of IL-13 and elevated numbers of goblet cells in the small intestine.34 This demonstrates that the observed functional effects of TL1A are independent of the cell type that produces TL1A in vivo. Although TL1A is produced as a type II transmembrane protein, it is also secreted as soluble protein from activated DCs,20 and therefore it is possible that the effects of TL1A in CD11c-TL1A tg mice could have been mediated by soluble TL1A released from DCs.
Constitutive expression of TL1A resulted in increased frequencies of CD4+ T cells with an effector/memory phenotype (Figure 3b), suggesting that these cells could represent the source for IL-13 and IL-17 in CD11c-TL1A tg mice. Examination of small intestinal CD4+ LPLs from CD11c-TL1A tg mice demonstrated that the frequencies of IL-13- and IL-17-producing CD4+ T cells were both increased when compared with LPLs from wild-type mice (Figure 4c). However, these increases were relatively small raising the possibility that the high levels of IL-13 in the small intestinal tissue were derived from another cell type. Invariant NKT cells that express DR3 could potentially contribute to elevated levels of IL-13 in the small intestine, as DR3 triggering in vitro was shown to costimulate IL-13 production by these cells.14 However, we observed fewer invariant NKT cells in CD11c-TL1A tg mice (Figure 3a), making their contribution to the increased levels of IL-13 in CD11c-TL1A tg mice unlikely. Generation of CD11c-TL1A tg mice on an invariant NKT cell-deficient background is required to definitively exclude a role for invariant NKT cells in this model. Other cell types that may contribute to enhanced IL-13 production in CD11c-TL1A tg mice include mast cells, basophils, and the recently identified nuocytes and natural helper cells.35, 36, 37 Future studies using IL-13 reporter mice35 may help elucidate other cell types involved in IL-13 production in CD11c-TL1A tg mice.
The data presented in this study showing that expression of TL1A under steady-state conditions preferentially increases the frequencies of Th2 and Th17 but not Th1 cells extend previous findings on the role of the TL1A–DR3 interaction in effector T-cell responses. Thus, in vitro-generated Th17 cells were shown to express more DR3 when compared with Th1 and Th2 cells, and this allowed them to be more responsive to TL1A-mediated costimulation in vitro.15 Furthermore, when CD4+ T cells are cultured under nonpolarizing conditions, the lack of DR3 signaling was shown to negatively impact the production of Th2 cytokines during the secondary response, but not IFN-γ.14 It should be noted, however, that in some settings DR3 signaling has been shown to influence IFN-γ-producing cells. Thus, in experimental autoimmune encephalomyelitis, the absence of TL1A or DR3 resulted in diminution of not only IL-17+ T cells, but also IFN-γ+ and IL-17/IFN-γ dual expressing T cells that infiltrate the spinal cord.15, 19 Th17 cells can diverge to a Th1 phenotype in response to IL-12,38 and therefore it is possible that TL1A could exert effects on IFN-γ production depending on the balance between Th1- and Th17-promoting cytokines. TL1A and DR3 are expressed in ulcerative colitis and Crohn's disease lesions,21 in which IL-13 and IL-17/IFN-γ, respectively, are implicated in disease pathogenesis.39, 40 Our findings suggest that the TL1A–DR3 interaction could exacerbate immunopathology in both of these conditions by either stimulating increased production of IL-13 or promoting Th17 responses.
An unexpected finding that was revealed by analysis of TL1A tg mice is the effect of TL1A on Treg cells. We demonstrate that triggering of DR3 on Treg cells can enhance their activation and turnover (Figures 5 and 6). Treg cells expand during the normal course of an immune response,6, 7, 41 and their ability to suppress is enhanced after proliferation.42 Our data suggest that expression of TL1A on DCs could contribute to the expansion of Treg cells, while at the same time attenuating their suppressive effects on conventional T cells (Figure 7). This will have the effect of allowing effector T cells to mount a protective immune response despite the presence of increasing numbers of Treg cells. Because of the transient nature of TL1A expression, Treg cells will be expected to re-gain their full suppressive capability relatively quickly and thus dampen the magnitude of the inflammatory response.6, 7, 41, 43, 44 Indeed, Treg cells isolated from CD11c-TL1A tg mice were highly efficient in suppressing the proliferation of effector T cells in vitro in the absence of exogenous soluble recombinant TL1A (sTL1A; data not shown). Using DR3-deficient or DR3-sufficient Treg cells and conventional CD4+ T cells, we were able to demonstrate that DR3 signaling in both cell types contributes to the ability of TL1A to overcome suppression (Figure 7). Whether DR3 exerts these effects through mechanisms that are shared with OX40, GITR, or IL-6, which have also been reported to reverse suppression,45 remains to be determined in future studies.
Methods
Mice, reagents, and analysis of gene expression. To generate CD11c-TL1A tg mice, a DNA fragment containing the coding sequence of TL1A was cloned into the EcoRI site of CD11c.pDOI-5.46 A NotI/XhoI digested fragment lacking vector sequences was microinjected into the male pronuclei of FVB/N zygotes. CD11c-TL1A tg mice were identified by PCR of genomic DNA using the following primers: 5′-CTCACCTCCCAGAGGCAAG-3′ and 5′-GGAGTCTGGCTTGTTTGGTCG-3′. Mice were backcrossed to the C57BL/6 strain for four to five generations. Transgenic mice and age-matched littermate controls were used at 6–24 weeks of age. sTL1A consisting of the extracellular domain of TL1A fused to the C-terminus of a rat CD4 domains 3 and 4 tag was produced in Chinese hamster ovary cells and purified by immunoaffinity column chromatography.47 Expression of TL1A, IL-17, IL-13, IL-4, and IFN-γ was determined by quantitative RT-PCR using TaqMan gene expression assays Mm00770031_m1, Mm00439619_m1, Mm00434204_m1, Mm00445260_m1, and Mm00801778_m1, respectively (Applied Biosystems, Warrington, UK). Fold expression of TL1A was calculated using the 2−ΔΔCt method and hypoxanthine-guanine phosphoribosyltransferase (TaqMan assay Mm00446968_m1) or β2-microglobulin (TaqMan assay Mm00437762_m1) as a reference gene. For relative expression analysis of cytokine genes, we first normalized the measurement to expression of a reference gene (ΔCt) and then used 2−ΔCt to obtain gene expression levels. Gene expression levels were then normalized to the average gene expression in wild-type mice. DR3-deficient and Foxp3–GFP knock-in mice were described previously.26, 48 Animal experiments were carried out according to the UK Home Office license guidelines and approved by the University of Southampton's ethical committee.
Immunofluorescence confocal microscopy. Frozen sections (10 μm) were fixed in acetone, blocked with 5% normal goat serum, and incubated with rat (IgG) anti-TL1A (Tan2.2; ref. 49) and hamster anti-CD11c (clone N418) followed by Alexa Fluor 546-conjugated goat anti-rat IgG and Alexa Fluor 488-conjugated goat anti-hamster IgG (Invitrogen, Paisley, UK). Sections were washed with phosphate-buffered saline and incubated with rabbit anti-CD3 (Abcam, Cambridge, UK) followed by Alexa Fluor 647-conjugated goat anti-rabbit IgG (Molecular Probes). Sections were mounted in Vectashield (Vector Laboratories, Burlingham, CA) and images collected sequentially on a Leica (Milton Keynes, UK) SP5 CLSM using Argon (488 nm), GreNe (543 nm), and HeNe (633 nm) lasers and a pinhole equivalent to 1 Airy disc. Image (TIFF) files were transferred to Adobe Photoshop (Adobe Systems Europe, Uxbridge, UK) and contrast stretched to use the whole gray scale.
Histology. Tissue was fixed in 10% neutral buffered formalin (Sigma, Poole, UK). Samples were embedded, sectioned, and stained with hematoxylin and eosin.
Isolation of LPLs. After washing with phosphate-buffered saline, small intestinal specimens were incubated in RPMI media containing 20 mM HEPES, antibiotics, 3% fetal calf serum, 5 mM EDTA, and 0.145 mg ml–1 dithiothreitol for 20 min at 37 °C with stirring. The tissue was then placed in media with antibiotics and 2 mM EDTA and agitated for 30 s. This procedure was repeated three more times before tissue digestion was performed for 15 min at 37 °C in RPMI media containing 20 mM HEPES, Liberase DL (200 μg ml–1), and DNAse I (0.5 mg ml–1). Fresh media containing enzymes was then added and intestinal tissue was digested for a further 15 min. The tissue was then mashed and the released cells passed sequentially through 100, 70, and finally 40 μm cell strainers.
T-cell proliferation and suppression assays. CD4+CD25+ and CD4+CD25− cells were purified to ≥95 and ≥98%, respectively, by magnetic selection (Miltenyi Biotec, Bergisch Gladbach, Germany). Carboxyfluorescein succinimidyl ester-labeled CD4+CD25− cells (1.25 × 105 per well) were cultured with Treg cells in the presence of irradiated DR3-deficient antigen-presenting cells (2.5 × 105 per well; 3000 rad) and anti-CD3 (145.2C11, 2 μg ml–1) or anti-CD3 and sTL1A (5 μg ml–1). The costimulatory effect of sTL1A on Treg cell proliferation was evaluated at 72 h by 3H-thymidine incorporation following stimulation with anti-CD3 or anti-CD3 and anti-CD28 (37.51, 5 μg ml–1) with or without sTL1A. For these assays, magnetically selected CD4+CD25+ T cells (2.5 × 104 cells per well) or flow-sorted Foxp3+GFP+ Treg cells (FACSAria II, ≥99% pure, BD Biosciences, Oxford, UK) were cultured in U-shaped 96-well plates with irradiated DR3-deficient splenocytes (5 × 104 per well).
Flow cytometry. Intracellular Foxp3 and Ki-67 staining was carried out using mAbs from eBioscience (Hatfield, UK) and BD Biosciences (Oxford, UK), respectively, and fixation/permeabilization reagents from eBioscience. For intracellular cytokine staining, cells (3 × 106 per ml) were stimulated for 16 h with anti-CD3 (2 μg ml–1) and anti-CD28 (5 μg ml–1). Brefeldin A (GolgiPlug, BD Biosciences, Oxford, UK) was added to the culture media for the last 4 h and cells were then stained for IL-17 (TC11-18H10.1), IFN-γ (XMG1.2), or IL-13 (eBio13A) using the Cytofix/Cytoperm kit from BD Pharmingen. CD1d tetramer was from Proimmune (Oxford, UK).
Statistical analysis. Error bars represent the s.e.m. Statistical significance was assessed using a two-tailed Student's t-test or Mann–Whitney test in the case of cytokine gene expression data using the GraphPad Prism software La Jolla, CA.
References
Xavier, R.J. & Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434 (2007).
Barnes, M.J. & Powrie, F. Regulatory T cells reinforce intestinal homeostasis. Immunity 31, 401–411 (2009).
Sakaguchi, S., Yamaguchi, T., Nomura, T. & Ono, M. Regulatory T cells and immune tolerance. Cell 133, 775–787 (2008).
Thornton, A.M. & Shevach, E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188, 287–296 (1998).
Fisson, S. et al. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J. Exp. Med. 198, 737–746 (2003).
Scott-Browne, J.P. et al. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J. Exp. Med. 204, 2159–2169 (2007).
Lund, J.M., Hsing, L., Pham, T.T. & Rudensky, A.Y. Coordination of early protective immunity to viral infection by regulatory T cells. Science 320, 1220–1224 (2008).
Bour-Jordan, H. & Bluestone, J.A. Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol. Rev. 229, 41–66 (2009).
van Olffen, R.W. et al. GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J. Immunol. 182, 7490–7500 (2009).
Takeda, I. et al. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J. Immunol. 172, 3580–3589 (2004).
Zheng, G., Wang, B. & Chen, A. The 4-1BB costimulation augments the proliferation of CD4+CD25+ regulatory T cells. J. Immunol. 173, 2428–2434 (2004).
Valzasina, B., Guiducci, C., Dislich, H., Killeen, N., Weinberg, A.D. & Colombo, M.P. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood 105, 2845–2851 (2005).
Stephens, G.L. et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J. Immunol. 173, 5008–5020 (2004).
Fang, L., Adkins, B., Deyev, V. & Podack, E.R. Essential role of TNF receptor superfamily 25 (TNFRSF25) in the development of allergic lung inflammation. J. Exp. Med. 205, 1037–1048 (2008).
Pappu, B.P. et al. TL1A-DR3 interaction regulates Th17 cell function and Th17-mediated autoimmune disease. J. Exp. Med. 205, 1049–1062 (2008).
Migone, T.S. et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity 16, 479–492 (2002).
Papadakis, K.A. et al. TL1A synergizes with IL-12 and IL-18 to enhance IFN-gamma production in human T cells and NK cells. J. Immunol. 172, 7002–7007 (2004).
Bamias, G. et al. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc. Natl. Acad. Sci. USA 103, 8441–8446 (2006).
Meylan, F. et al. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity 29, 79–89 (2008).
Prehn, J.L., Thomas, L.S., Landers, C.J., Yu, Q.T., Michelsen, K.S. & Targan, S.R. The T cell costimulator TL1A is induced by FcgammaR signaling in human monocytes and dendritic cells. J. Immunol. 178, 4033–4038 (2007).
Bamias, G. et al. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J. Immunol. 171, 4868–4874 (2003).
McKenzie, G.J., Bancroft, A., Grencis, R.K. & McKenzie, A.N. A distinct role for interleukin-13 in Th2-cell-mediated immune responses. Curr. Biol. 8, 339–342 (1998).
Steenwinckel, V. et al. IL-9 promotes IL-13-dependent paneth cell hyperplasia and up-regulation of innate immunity mediators in intestinal mucosa. J. Immunol. 182, 4737–4743 (2009).
Minty, A. et al. The related cytokines interleukin-13 and interleukin-4 are distinguished by differential production and differential effects on T lymphocytes. Eur. Cytokine Netw. 8, 203–213 (1997).
Heller, F., Fuss, I.J., Nieuwenhuis, E.E., Blumberg, R.S. & Strober, W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity 17, 629–638 (2002).
Bettelli, E. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006).
Ramalingam, T.R. et al. Unique functions of the type II interleukin 4 receptor identified in mice lacking the interleukin 13 receptor alpha1 chain. Nat. Immunol. 9, 25–33 (2008).
Junttila, I.S. et al. Tuning sensitivity to IL-4 and IL-13: differential expression of IL-4Ralpha, IL-13Ralpha1, and gammac regulates relative cytokine sensitivity. J. Exp. Med. 205, 2595–2608 (2008).
Herbert, D.R. et al. Intestinal epithelial cell secretion of RELM-beta protects against gastrointestinal worm infection. J. Exp. Med. 206, 2947–2957 (2009).
Kontoyiannis, D. et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn's-like inflammatory bowel disease. J. Exp. Med. 196, 1563–1574 (2002).
Shaikh, R.B. et al. Constitutive expression of LIGHT on T cells leads to lymphocyte activation, inflammation, and tissue destruction. J. Immunol. 167, 6330–6337 (2001).
Arens, R. et al. Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNgamma-mediated B cell depletion. Immunity 15, 801–812 (2001).
Murata, K., Nose, M., Ndhlovu, L.C., Sato, T., Sugamura, K. & Ishii, N. Constitutive OX40/OX40 ligand interaction induces autoimmune-like diseases. J. Immunol. 169, 4628–4636 (2002).
Meylan, F. et al. The TNF-family cytokine TL1A drives IL-13-dependent small intestinal inflammation. Mucosal Immunol (in press).
Neill, D.R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Moro, K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Eberl, G. Immunology: close encounters of the second type. Nature 464, 1285–1286 (2010).
Lee, Y.K. et al. Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107 (2009).
Strober, W., Fuss, I. & Mannon, P. The fundamental basis of inflammatory bowel disease. J. Clin. Invest. 117, 514–521 (2007).
Maynard, C.L. & Weaver, C.T. Intestinal effector T cells in health and disease. Immunity 31, 389–400 (2009).
Haribhai, D., Lin, W., Relland, L.M., Truong, N., Williams, C.B. & Chatila, T.A. Regulatory T cells dynamically control the primary immune response to foreign antigen. J. Immunol. 178, 2961–2972 (2007).
Gavin, M.A., Clarke, S.R., Negrou, E., Gallegos, A. & Rudensky, A. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat. Immunol. 3, 33–41 (2002).
McGeachy, M.J., Stephens, L.A. & Anderton, S.M. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J. Immunol. 175, 3025–3032 (2005).
Leech, M.D., Benson, R.A., De Vries, A., Fitch, P.M. & Howie, S.E. Resolution of Der p1-induced allergic airway inflammation is dependent on CD4+CD25+Foxp3+ regulatory cells. J. Immunol. 179, 7050–7058 (2007).
Shevach, E.M. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645 (2009).
Brocker, T., Riedinger, M. & Karjalainen, K. Targeted expression of major histocompatibility complex (MHC) class II molecules demonstrates that dendritic cells can induce negative but not positive selection of thymocytes in vivo. J. Exp. Med. 185, 541–550 (1997).
Al-Shamkhani, A., Mallett, S., Brown, M.H., James, W. & Barclay, A.N. Affinity and kinetics of the interaction between soluble trimeric OX40 ligand, a member of the tumor necrosis factor superfamily, and its receptor OX40 on activated T cells. J. Biol. Chem. 272, 5275–5282 (1997).
Wang, E.C., Thern, A., Denzel, A., Kitson, J., Farrow, S.N. & Owen, M.J. DR3 regulates negative selection during thymocyte development. Mol. Cell. Biol. 21, 3451–3461 (2001).
Bull, M.J. et al. The death receptor 3-TNF-like protein 1A pathway drives adverse bone pathology in inflammatory arthritis. J. Exp. Med. 205, 2457–2464 (2008).
Acknowledgements
We are grateful to Nick Jones (University of Oxford) and Terry Strom (Harvard Medical School) for providing Foxp3–GFP knock-in mice. A. Al-S is in receipt of grants from Cancer Research UK, the Association for International Cancer Research, Leukaemia & Lymphoma Research, and the University of Southampton.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article
Cite this article
Taraban, V., Slebioda, T., Willoughby, J. et al. Sustained TL1A expression modulates effector and regulatory T-cell responses and drives intestinal goblet cell hyperplasia. Mucosal Immunol 4, 186–196 (2011). https://doi.org/10.1038/mi.2010.70
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.70
This article is cited by
-
Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome
Mucosal Immunology (2018)
-
TNF superfamily cytokines in the promotion of Th9 differentiation and immunopathology
Seminars in Immunopathology (2017)
-
TL1A/DR3 axis involvement in the inflammatory cytokine network during pulmonary sarcoidosis
Clinical and Molecular Allergy (2015)
-
A major population of mucosal memory CD4+ T cells, coexpressing IL-18Rα and DR3, display innate lymphocyte functionality
Mucosal Immunology (2015)
-
TNF superfamily member TL1A elicits type 2 innate lymphoid cells at mucosal barriers
Mucosal Immunology (2014)
