
Abstract
A critical function of the intestinal mucosa is to form a barrier that separates luminal contents from the interstitium. This intestinal barrier is compromised in a number of intestinal diseases, most notably inflammatory bowel disease. In vitro studies have demonstrated that cytokines elaborated by immune cells can cause the mucosal barrier to become leaky; these cytokines are known to be increased in intestinal mucosa involved in inflammatory bowel disease. Detailed information describing the mechanisms by which altered cytokine signaling occurs is not available, but recent data implicate the cytoskeleton within epithelial cells as a critical regulator of the mucosal barrier under physiological and pathophysiological conditions. Using available data, we describe a model of intestinal disease where an initial insult to the epithelial barrier may trigger a self-amplifying cycle of immune activation, cytokine release, and further barrier dysfunction. This model is supported by the observation that pharmacological abrogation of cytokine signaling corrects both barrier defects and clinical disease in animal models and human patients, although such therapy clearly has multiple mechanisms. Other therapeutic targets that represent strategies to prevent or reverse disease processes are also considered. The overarching hypothesis is that modulation of the mucosal epithelial barrier plays a critical role in the initiation and propogation of inflammatory intestinal diseases.
Similar content being viewed by others

Main
Crohn's disease and ulcerative colitis are chronic disorders of the intestines, collectively known as inflammatory bowel disease. It is estimated that up to one million Americans suffer from inflammatory bowel disease, with approximately 30 000 new cases diagnosed each year. The peak age of diagnosis is between 15 and 35 years, and more than 10% of new presentations occur in patients less than 18 years of age. Given the young age of presentation of these chronic illnesses, the subsequent lifetime morbidity is substantial. For example, the direct and indirect costs of inflammatory bowel disease in 2000 have been estimated to be in excess of $1.2 billion.1 Inflammatory bowel disease has a familial link; approximately 20% of patients have a relative with either Crohn's disease or ulcerative colitis and several inflammatory bowel disease-related genes have been identified.2, 3, 4 Thus, although the causes of inflammatory bowel disease remain unknown, a variety of epidemiologic, genetic, morphologic, and biochemical data provide some clues as to mechanisms and pathogenesis.
While these diseases are often viewed as a single entity with common symptoms, differing morphologies and disease courses demonstrate that these are distinct diseases that can be separately classified in the majority of patients. For example, while both diseases show characteristic features of chronic mucosal damage, including epithelial metaplasia, glandular atrophy, and architectural distortion, the presence of fissuring ulcers or fistula tracts, strictures, deep granulomas, skip lesions, or small intestinal disease all result in the classification of a patient as having Crohn's disease. In contrast, while backwash ileitis may occasionally be present, the small intestine is typically normal in ulcerative colitis, disease is continuous from the rectum to a proximal sharp demarcation of disease, and strictures, fistulae, and granulomas should not be present. However, classification is not always straightforward. For example, cases of inflammatory bowel disease with left-sided disease and involvement of cecal/periappendiceal mucosa or the appendix itself are recognized to be ulcerative colitis, despite the potential for interpretation as a skip lesion.5, 6, 7 The subset of approximately 10% of inflammatory bowel disease cases that defy absolute classification as either Crohn's disease or ulcerative colitis further highlights the tremendous clinical, morphological, and biological overlap between Crohn's disease and ulcerative colitis.
A role for the immune system in inflammatory bowel disease is obvious. Indeed, in murine models, adoptive transfer of CD45RB high T cells induces an inflammatory intestinal disease with many features of inflammatory bowel disease, including granulomas, mucosal architectural distortion, crypt abscesses, and lamina propria mononuclear infiltrates.8 However, the available data suggest that epithelial dysfunction may have an equally important role in the disease process. For example, numerous in vivo human studies have shown that disruption of intestinal epithelial barrier function closely mirrors disease activity and actually correlates with CD45RO expression on circulating CD19 positive B cells.9, 10, 11 Even more intriguing is the observation that, in clinically asymptomatic Crohn's disease patients, increased intestinal epithelial permeability precedes clinical relapse by as much as 1 year,12, 13, 14 indicating that a permeability defect may be an early event in disease reactivation. Further evidence supporting the hypothesis that abnormal intestinal barrier function occurs early in the pathogenesis of Crohn's disease comes from numerous studies showing that a subset of clinically healthy first-degree relatives of Crohn's disease patients have abnormally increased intestinal permeability.10, 15, 16, 17, 18, 19, 20 Notably, the presence of this permeability defect in genetically unrelated relatives, for example, spouses, suggests that this abnormal permeability may be secondary to environmental as well as genetic factors.17 Nonetheless, the potential importance of this permeability defect is emphasized by a case report of a healthy first-degree relative of Crohn's disease patients who exhibited increased intestinal permeability at the age of 13 years. She was evaluated thoroughly, including biopsies, small bowel followthrough, and 111Indium-labeled white cell scan and showed no clinical features of intestinal disease.21 However, 8 years later, at the age of 21 years, the subject returned with stricturing and ulcerated ileocolonic Crohn's disease.21 Although a single case report, this patient shows that a permeability defect can exist long before the onset of full-blown disease, indicating that, at least in this case, an intestinal permeability defect may have been an early event in disease pathogenesis.
Increased intestinal permeability is not unique to inflammatory bowel disease; several other diseases, not all of which include a significant inflammatory component, also exhibit altered permeability. Graft vs host disease, as occurs following bone marrow transplantation, may be the best example of this. Donor T cells react to host tissues and produce diseases ranging from severe, widespread tissue destruction to diarrhea. This diarrhea is associated with acute epithelial damage, including pronounced crypt cell apoptosis, and can proceed to a chronic injury pattern with glandular atrophy. Active graft vs host disease is also associated with a significant intestinal permeability defect.22 Although this permeability defect could conceivably be due to the marked apoptosis that occurs, numerous studies have now clearly shown that epithelial cell apoptosis alone is insufficient to cause permeability deficits.23, 24, 25, 26
Permeability defects associated with malabsorption are also prominent in celiac sprue along with the classic histological features of villous blunting and increased numbers of intraepithelial lymphocytes.27, 28, 29 At first this seems counterintuitive, since villous atrophy leads to decreased epithelial surface area and should therefore reduce paracellular permeability. Moreover, as ulceration and erosion are distinctly unusual in celiac disease, the increases in permeability cannot be explained as a result of gross epithelial loss. Thus, the permeability defects present in sprue are best understood as the result of epithelial dysfunction rather than epithelial destruction. Similarly, enteric bacterial and parasitic infections are known to result in barrier defects.30, 31 These have been studied in great detail using in vitro systems and, in the case of enteropathogenic Escherichia coli, depend on the presence of specific bacterial proteins that communicate with the epithelial cell.32 In the cases of Giardia lamblia infection, the increases in permeability are also not due to direct tissue damage or tissue invasion by microorganisms, since neither is evident histologically. However, as discussed below, the intestinal permeability defect induced by Giardia lamblia infection does appear to require the activation of specific signal transduction pathways within host intestinal epithelial cells.33, 34 Thus, an understanding of the regulation of the epithelial barrier and its interaction with a variety of host- and pathogen-derived extracellular signals may yield insight into a wide array of intestinal diseases.
Paracellular permeability and barrier function are primarily determined by the epithelial tight junction
As implied above, the gastrointestinal epithelium forms a barrier that separates the finely regulated homeostasis of the body interstitium from the harsh environment of the intestinal lumen. The intestinal epithelial cell plasma membrane serves as an effective barrier to most hydrophilic solutes. However, the paracellular space must also be sealed to form an intact epithelial barrier. This seal is provided by the tight junction.35 Tight junctions cannot be well visualized by light microscopy. However, their location is easily resolved on hematoxylin and eosin-stained slides as the terminal bar, the refractile area of membrane and cytoplasm just subapical to the brush border. Transmission electron microscopy demonstrates the tight junction to be a discrete region of membrane apposition between adjacent epithelial cells at the luminal aspect of the apical junction complex. At this site, the adjacent plasma membranes appear to fuse.36, 37, 38 This led to the initial misinterpretation of the tight junction as an impermeable barrier.39
As further studies showed the tight junction to be the rate-limiting step in the paracellular pathway, it became apparent that the tight junction forms a selectively permeable barrier. The barrier exhibits both size and charge selectivity, a vital attribute for the regulation of fluid and solute movement. Indeed, these barrier functions differ remarkably between tissues. For example, the mammalian small intestinal paracellular pathway is at least four-fold more permeable to K+ than to Cl− ions, while this selectivity is altered significantly in other areas of the gastrointestinal tract. The overall permeability also varies more than 30-fold, depending on the tissue type studied.40, 41 Freeze fracture electron microscopy shows the tight junction to be a series of anastomosing strands. Mathematical analyses suggested that the strands did not function as resistors in series, but housed a series of channels with individual open and closed probabilities.42
The precise roles of many tight junction proteins remain unknown. Numerous transmembrane proteins, such as claudins and occludin,43, 44 and cytoplasmic peripheral membrane proteins, including ZO-1, -2, and -3, and cingulin,45, 46, 47, 48, 49 have been described. Although recent work has clarified the roles of the claudin family of proteins, the specific roles of others remains enigmatic. It appears that claudins form the actual pores that determine the charge selectivity of the paracellular pathway.50, 51, 52 For example, human mutations in paracellin-1/claudin-16 result in defective paracellular reabsorption of Mg+2 across the renal proximal tubule.53 This results in renal Mg+2 wasting and a familial hypomagnesaemia syndrome that cannot be corrected by Mg+2 supplementation.53, 54 Thus, mutation of a single protein can have enormous consequences for the permeability of the tight junction complex.
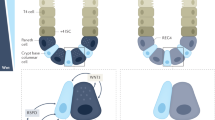
The tight junction protein complex is intimately related to the apical perijunctional actomyosin ring: functionally, structurally, and biochemically (Figure 1). Many tight junction proteins interact with both F-actin and myosin.45, 46, 55, 56 These interconnections between the various tight junction proteins and cytoskeletal elements are believed to stabilize the tight junction and to be critical to its regulation.57

The tight junction. Although the tight junction cannot be seen on hematoxylin- and eosin-stained sections, immunofluorescence demonstrates the restricted location of the tight junction (top inset). Here, nuclei are stained blue, actin is green, and ZO-1, a tight junction protein, is red. Virtually all of the ZO-1 colocalizes with perijunctional actin, producing a yellow color at the tight junction. A simplified schematic of the tight junction is also shown (bottom inset). The claudin family of proteins forms the actual paracellular pore within the tight junction and is associated with another transmembrane protein, occludin. ZO-1 and other cytoplasmic proteins, such as ZO-2 and ZO-3, attach to this complex. Several of these proteins, including occludin and ZO-1, interact directly with F-actin.
Tight junction permeability is plastic
Although previous models of the tight junction considered it to have static permeability properties, more recent research has shown it to be a dynamic structure with the ability to alter its permeability in response to extracellular stimuli. Physiologically, the response of the tight junction to luminal glucose is particularly well studied. Transcellular transport of glucose, predominantly under the control of the Na+–glucose cotransporter SGLT1, is a saturable system. However, the transport of glucose across the epithelial barrier is not saturable; continued increases in luminal glucose concentration result in continued increases in glucose absorption.58 These data suggest that an alternative diffusion-driven pathway for glucose transport exists. The existence of this pathway has been confirmed in studies in humans and experimental animals,59, 60 isolated human or rodent small intestinal mucosa,25, 61, 62, 63 and intestinal epithelial cell lines.62, 64 These studies all confirm an increase in paracellular permeability in response to Na+–glucose cotransport. In isolated tissues and cell lines, this corresponds to a decrease in transepithelial resistance, an inverse measure of paracellular permeability. Transmission electron microscopy of isolated rodent mucosa showed that Na+–glucose cotransport was accompanied by condensation of microfilaments within the perijunctional actomyosin ring, suggesting actomyosin contraction. Subsequent analyses using cell lines and isolated human and rodent small intestine showed that this condensation was driven by myosin light chain kinase-mediated phosphorylation of myosin II regulatory light chain.62, 63 This myosin light chain phosphorylation occurs within the perijunctional actomyosin ring and colocalizes with the tight junction.63 Moreover, inhibition of myosin light chain kinase blocks both Na+–glucose cotransport-induced myosin light chain phosphorylation and tight junction regulation in isolated mucosa and intestinal epithelial cell lines.62, 63, 65 Consistent with this central role of myosin light chain phosphorylation in regulating paracellular permeability, studies using an inducible constitutively active myosin light chain kinase have shown that this activity is sufficient to activate the downstream events necessary for tight junction regulation.57 Although this is only one physiological mechanism of tight junction regulation, accumulating evidence suggests that myosin light chain kinase-mediated regulation of tight junction permeability is a common intermediate in a variety of physiological and pathophysiological pathways related to altered paracellular permeability in vitro and in vivo.57, 66, 67
Bugs, cytokines, and drugs: factors that influence the tight junction barrier
The spectrum of barrier dysfunction that occurs in patients with intestinal disease suggests that pathophysiological factors may hijack the normal physiological pathways that regulate tight junction permeability. Consistent with this hypothesis, enteropathogenic E. coli (EPEC) infection causes diarrhea in pediatric patients and an in vitro model of this infection using intestinal epithelial cell lines demonstrates that EPEC induce a large increase in tight junction permeability. This is accompanied by disruption of tight junction morphology, including reorganization of the actin cytoskeleton, redistribution of tight junction proteins, and increased myosin light chain phosphorylation and can be reversed by inhibition of myosin light chain kinase.24, 68 Thus, the data suggest that EPEC utilizes an intrinsic pathway of tight junction regulation through myosin light chain kinase to affect barrier dysfunction. Similarly, the barrier disruption induced by Giardia infection can also be reversed by myosin light chain kinase inhibition, suggesting that, like EPEC, Giardia also disrupts tight junction permeability via myosin light chain phosphorylation.34
Increased epithelial permeability is not only caused by exogenous factors such as infection; a growing body of evidence suggests that the immune system plays an important role in modulating intestinal permeability. Two cytokines, interferon-γ (IFNγ) and tumor necrosis factor-α (TNFα), are found in high levels in intestinal mucosa involved in inflammatory bowel disease.69, 70 These same two cytokines have also been found to decrease barrier function of cultured intestinal epithelial monolayers.24, 71, 72, 73 Incubation of intestinal epithelial cell monolayers with both IFNγ and TNFα leads to reorganization of many tight junction proteins, including ZO-1, junctional adhesion molecule 1, occludin, claudin-1, and claudin-4.23 The changes in paracellular permeability caused by IFNγ and TNFα are associated with marked increases in myosin light chain phosphorylation and can be reversed using a specific membrane permeant inhibitor of myosin light chain kinase, indicating that these cytokines also utilize the myosin light chain kinase-driven pathway to increase tight junction permeability.24 Thus, a key step in the pathogenesis of inflammatory bowel disease may be myosin light chain kinase activation by IFNγ and TNFα, leading to intestinal barrier dysfunction. Experiments using animal models of inflammatory bowel disease can provide an insight into the origin of these cytokines in the disease process as well as further elucidate the interactions of the immune system with the intestinal epithelium.
What can animal models teach us about human disease?
Over 60 different animal models of inflammatory bowel disease have been reported74 and there are numerous excellent models of graft vs host disease. While no single animal model is a perfect replica of human disease, certain features are common to many models, indicating that these principles may also apply to human disease. For example, almost all animal inflammatory bowel disease models require the presence of intestinal flora; animals raised in a germ-free environment are rarely affected. This highlights the likely importance of exogenous stimuli in the development and persistence of the abnormal immune response observed in intestinal disease. While the exact factor(s) involved in this process are unknown, studies of a murine graft vs host disease model have shown that lipopolysaccharide sensitivity can predict disease severity and that lipopolysaccharide antagonism can inhibit disease progression.75, 76
The IL-10 knockout mouse model of intestinal disease demonstrates two other commonalities of animal intestinal disease models: the importance of the genetic background of the animal and the necessity of T cells and their interactions with macrophages in the TH1 immune response. The loss of IL-10 removes a key mechanism downregulating macrophage activation by T cells, leading to an unchecked TH1 inflammatory response.77 This response is associated with an intestinal permeability defect upon exposure to intestinal flora.78 Along with other genetic inflammatory bowel disease models, this demonstrates the necessity of activated macrophages, T cells, and cytokines associated with the TH1 response in promoting inflammatory changes and altering the intestinal barrier. This inflammation is also subject to genetic modulation. For example, the IL-10 knockout produces a severe colitis on a C3H genetic background, while C57Bl/6J mice lacking IL-10 only develop mild disease.79 Thus, although the exact genes involved are unknown, other genetic differences between inbred laboratory mouse strains must contribute to the development and severity of intestinal disease.
Almost all animal models of inflammatory bowel disease involve disruption or abnormal stimulation of the immune system, such as T-cell transfer, genetic disruption of the immune system, or chemical stimulation of inflammation. However, one model exists in which a selective disruption of epithelial function leads to an inflammatory disease involving the intestines.80 In this model, disruption of the adhesion molecule E-cadherin by tissue-specific expression of a dominant negative cadherin construct in small intestinal epithelial cells throughout the crypt-villus axis resulted in disruption of adherens junctions. When chimeric mice were created, it was evident that a defect in cell migration and proliferation only occurred in epithelial units expressing the dominant negative cadherin.80 By 3 months of age, the mice developed typical histological features of inflammatory bowel disease, including architectural distortion, crypt abscesses, and both aphthous and linear ulcers.80 Strikingly, in chimeric mice, these changes were limited to epithelia expressing dominant negative cadherin.80 Thus, this model indicates that epithelial dysfunction alone may be sufficient to initiate the disease process in the absence of systemic inflammatory disease.
Interdependence of inflammation and barrier function in intestinal disease
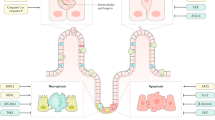
The accumulated evidence from studies of patients, animals, and cultured cell models is sufficient to begin to construct a model of the pathogenesis of inflammatory diseases of the intestine (Table 1). Three key components appear to be necessary in the progression of disease: (i) disruption of the epithelial barrier, (ii) access of luminal contents to the lamina propria, that is, immune cells, and (iii) an abnormal immune response (Figure 2). According to this model, a defect in the intestinal barrier allows contents of the intestinal lumen to mix freely with the contents of the lamina propria. Most importantly, these luminal contents include bacteria, bacterial products, food antigens, and other immunostimulatory antigens. Antigen presenting cells in the lamina propria process and present these antigens to T cells and also secrete IL-12, thereby directing the T cells to initiate a TH1 immune response. Central to the TH1 response is the secretion of IFNγ from the T cell, which activates macrophages to respond to the stimulus. During a normal TH1 response, antigen presenting cells also secrete IL-10, which acts to limit the TH1 response. This and other regulatory mechanisms may be disrupted in inflammatory bowel disease, allowing an abnormally robust inflammatory response. A central downstream event in this immune cascade is the secretion of TNFα from the activated macrophage. Two major cytokines, IFNγ and TNFα, then act on the epithelium, further disrupting the barrier and increasing permeability. In this manner, a vicious cycle is created in which barrier dysfunction allows further leakage of luminal contents, thereby triggering an immune response that can in turn feed back on the intestinal barrier to promote further leakiness. As various mouse models so clearly demonstrate, any one of these disease components, barrier dysfunction, abnormal immune stimulation, or an abnormal immune response, may initiate the cycle. This may occur as a genetic predisposition to a leaky barrier, an abnormal immune response, or environmental factors, such as infection, that cause immune stimulation. However, human inflammatory bowel disease shows abnormalities of all three components. Thus, effective therapy that blocks this cycle at one or more points may be effective in allowing the intestine to return to normal function.

A model for the pathogenesis of inflammatory bowel disease. (1) Initial barrier disruption may be caused by injury (ischemia, infection), genetic predisposition, or by underlying inflammation. This leads to a mixing (2) of luminal contents, including bacteria and other pathogens, with lamina propria contents, notably antigen presenting cells. These antigen presenting cells (3) process and present antigens in association with MHC class II molecules while simultaneously secreting cytokines such as IL-12 that promote a TH1 response. T cells (4) recognize the presented antigens and respond to the cytokine stimulus by secreting IFNγ to initiate the TH1 response. Simultaneously, loss or downregulation of anti-TH1 cytokines such as IL-10 (5) allows the inflammatory reaction to grow large and persist longer than usual. The IFNγ secreted by T cells promotes macrophage activation (6); these activated macrophages in turn secrete TNFα to promote the inflammatory reaction. TNFα and IFNγ also influence the epithelial barrier (7). Through unknown mechanisms, these cytokines activate MLCK (8), leading to MLC phosphorylation, actomyosin contraction, and opening of the tight junction. This leads to further loss of barrier function, continuing the cycle of disease progression.
Breaking the cycle: current and potential therapies for inflammatory bowel disease
For many years, the mainstay of therapy for active inflammatory bowel disease was corticosteroids. As a potent immunosuppressive, this therapy was effective in controlling disease; however, the severe side effects associated with corticosteroids drove the search for better therapies. Unfortunately, immunosuppression remains the core approach for medical treatment of active ulcerative colitis. However, the arrival of infliximab, the first therapy targeted towards a specific part of the disease cycle, has revolutionized the treatment of Crohn's disease. Continued advances in our understanding of inflammatory bowel disease offers several potential targets for further therapeutic breakthroughs.
TNFα: A current mainstay in the treatment of Crohn's disease is the monoclonal antibody infliximab. This anti-TNFα antibody downregulates the inflammatory process in Crohn's disease.81 Infliximab also induces apoptosis of lamina propria lymphocytes, decreases mucosal IFNγ production, and restores intestinal barrier function.82, 83, 84 Perhaps most striking is the observation that, in addition to reducing mucosal lymphocytic and neutrophilic infiltrates, mucosal architecture is restored in a subset of patients.81 Thus, in conjunction with its remarkable clinical therapeutic effect, infliximab normalizes histopathology, mucosal inflammation, cytokine production, and intestinal permeability. In a murine model of graft versus host disease, anti-TNFα therapy also normalizes mucosal histology, restores the mucosal barrier, and reduces serum lipopolysaccharide levels.75, 85 Thus, by potentially blocking several critical steps, including signaling between immune cells and from immune cells to the epithelium, inhibition of TNFα signaling corrects multiple aspects of intestinal disease.
IL-10: The presence of such striking intestinal disease in IL-10 knockout mice has spurred research into the possibility of using this cytokine as a therapy in human inflammatory bowel disease. Two studies have shown IL-10 to be moderately effective in the treatment of Crohn's disease.86, 87 In addition to its role in suppressing the TH1 immune response, IL-10 also appears to have a role in preserving the epithelial barrier of cultured cells in the face of IFNγ treatment, indicating that, like infliximab, the effects of IL-10 may extend beyond TH1 cells and macrophages.88
Myosin light chain kinase: As described above, myosin light chain kinase-mediated phosphorylation of myosin light chain is a central event in one pathway of tight junction regulation.24, 34, 55, 62, 63, 89 Moreover, myosin light chain kinase inhibition can correct barrier disruption induced in model intestinal epithelia by IFNγ and TNFα.24 Thus, myosin light chain kinase inhibition may provide a novel mechanism for restoration of intestinal barrier function that would stop the cycle of disease progression. While only a hypothesis at present, the success of myosin light chain kinase inhibitors in this and other in vitro and in vivo models suggests that this may be a therapy for the future.24, 34, 67
Unanswered questions
Despite significant progress in our understanding and treatment of inflammatory bowel disease and related intestinal disorders, numerous gaps in our knowledge remain. The initial events leading to the development of intestinal inflammatory diseases remain unclear. The identification of disease-associated mutations represents and an important first step in identifying the genetic factors that predispose individuals to the development of inflammatory bowel disease. However, it is likely that further mutations in proteins or regulatory pathways that control epithelial barrier function and the immune response remain to be discovered. In addition, evidence for environmental factors in disease pathogenesis suggests that further research into pathogens and other environmental factors may yield new insights into the development of inflammatory bowel disease. The factors that drive the continued abnormal immune responses are also undefined. Finally, the mechanisms by which the immune system leads to barrier dysfunction may be a particularly fertile area of future exploration. Although both TNFα and IFNγ can cause barrier dysfunction, the signaling pathways by which they increase paracellular permeability remain unknown. Elucidation of these pathways may yield further treatments for inflammatory bowel disease as well as related disorders, including graft vs host disease and enteric infections.
References
Sandler RS, Everhart JE, Donowitz M, et al. The burden of selected digestive diseases in the United States. Gastroenterology 2002;122:1500–1511.
Brant SR, Panhuysen CI, Nicolae D, et al. MDR1 Ala893 Polymorphism Is Associated with Inflammatory Bowel Disease. Am J Hum Genet 2003;73:1282–1292.
Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001;411:603–606.
Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 2001;411:599–603.
D'Haens G, Geboes K, Peeters M, et al. Patchy cecal inflammation associated with distal ulcerative colitis: a prospective endoscopic study. Am J Gastroenterol 1997;92:1275–1279.
Kroft SH, Stryker SJ, Rao MS . Appendiceal involvement as a skip lesion in ulcerative colitis. Mod Pathol 1994;7:912–914.
Goldblum JR, Appelman HD . Appendiceal involvement in ulcerative colitis. Mod Pathol 1992;5:607–610.
Leach MW, Bean AG, Mauze S, et al. Inflammatory bowel disease in C.B-17 scid mice reconstituted with the CD45RBhigh subset of CD4+ T cells. Am J Pathol 1996;148:1503–1515.
Casellas F, Aguade S, Soriano B, et al. Intestinal permeability to 99mTc-diethylenetriaminopentaacetic acid in inflammatory bowel disease. Am J Gastroenterol 1986;81:767–770.
Murphy MS, Eastham EJ, Nelson R, et al. Intestinal permeability in Crohn's disease. Arch Dis Child 1989;64:321–325.
Miki K, Moore DJ, Butler RN, et al. The sugar permeability test reflects disease activity in children and adolescents with inflammatory bowel disease. J Pediatr 1998;133:750–754.
Wyatt J, Vogelsang H, Hubl W, et al. Intestinal permeability and the prediction of relapse in Crohn's disease. Lancet 1993;341:1437–1439.
D'Inca R, Di Leo V, Corrao G, et al. Intestinal permeability test as a predictor of clinical course in Crohn's disease. Am J Gastroenterol 1999;94:2956–2960.
Arnott ID, Kingstone K, Ghosh S . Abnormal intestinal permeability predicts relapse in inactive Crohn disease. Scand J Gastroenterol 2000;35:1163–1169.
Yacyshyn BR, Meddings JB . CD45RO expression on circulating CD19+ B cells in Crohn's disease correlates with intestinal permeability. Gastroenterology 1995;108:132–137.
Munkholm P, Langholz E, Hollander D, et al. Intestinal permeability in patients with Crohn's disease and ulcerative colitis and their first degree relatives. Gut 1994;35:68–72.
Peeters M, Geypens B, Claus D, et al. Clustering of increased small intestinal permeability in families with Crohn's disease. Gastroenterology 1997;113:802–807.
Teahon K, Smethurst P, Levi AJ, et al. Intestinal permeability in patients with Crohn's disease and their first degree relatives. Gut 1992;33:320–323.
Hollander D . Permeability in Crohn's disease: altered barrier functions in healthy relatives? Gastroenterology 1993;104:1848–1851.
Katz KD, Hollander D, Vadheim CM, et al. Intestinal permeability in patients with Crohn's disease and their healthy relatives. Gastroenterology 1989;97:927–931.
Irvine EJ, Marshall JK . Increased intestinal permeability precedes the onset of Crohn's disease in a subject with familial risk. Gastroenterology 2000;119:1740–1744.
Johansson JE, Brune M, Ekman T . The gut mucosa barrier is preserved during allogeneic, haemopoietic stem cell transplantation with reduced intensity conditioning. Bone Marrow Transplant 2001;28:737–742.
Bruewer M, Luegering A, Kucharzik T, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol 2003;171:6164–6172.
Zolotarevsky Y, Hecht G, Koutsouris A, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002;123:163–172.
Madara JL . Maintenance of the macromolecular barrier at cell extrusion sites in intestinal epithelium: physiological rearrangement of tight junctions. J Membr Biol 1990;116:177–184.
Rosenblatt J, Raff MC, Cramer LP . An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol 2001;11:1847–1857.
Pearson AD, Eastham EJ, Laker MF, et al. Intestinal permeability in children with Crohn's disease and coeliac disease. Br Med J (Clin Res Ed) 1982;285:20–21.
Bjarnason I, Marsh MN, Price A, et al. Intestinal permeability in patients with coeliac disease and dermatitis herpetiformis. Gut 1985;26:1214–1219.
Greco L, D'Adamo G, Truscelli A, et al. Intestinal permeability after single dose gluten challenge in coeliac disease. Arch Dis Child 1991;66:870–872.
Johansen K, Stintzing G, Magnusson KE, et al. Intestinal permeability assessed with polyethylene glycols in children with diarrhea due to rotavirus and common bacterial pathogens in a developing community. J Pediatr Gastroenterol Nutr 1989;9:307–313.
Serrander R, Magnusson KE, Sundqvist T . Acute infections with Giardia lamblia and rotavirus decrease intestinal permeability to low-molecular weight polyethylene glycols (PEG 400). Scand J Infect Dis 1984;16:339–344.
McNamara BP, Koutsouris A, O'Connell CB, et al. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J Clin Invest 2001;107:621–629.
Buret AG, Mitchell K, Muench DG, et al. Giardia lamblia disrupts tight junctional ZO-1 and increases permeability in non-transformed human small intestinal epithelial monolayers: effects of epidermal growth factor. Parasitology 2002;125:11–19.
Scott KG, Meddings JB, Kirk DR, et al. Intestinal infection with Giardia spp. reduces epithelial barrier function in a myosin light chain kinase-dependent fashion. Gastroenterology 2002;123:1179–1190.
Fawcett DW . Intercellular bridges. Exp Cell Res 1961;(Suppl 8):174.
Robertson JD . The molecular structure and contact relationships of cell membranes. Prog Biophys Mol Biol 1960;10:343–418.
Farquhar MG, Palade GE . Junctional complexes in various epithelia. J Cell Biol 1963;17:375–412.
Moe H . The ultrastructure of Brunner's glands of the cat. J Ultrastruct Res 1960;4:58–72.
Kachar B, Reese TS . Evidence for the lipidic nature of tight junction strands. Nature 1982;296:464–466.
Diamond JM, Bossert WH . Standing-gradient osmotic flow. A mechanism for coupling of water and solute transport in epithelia. J Gen Physiol 1967;50:2061–2083.
Diamond JM, Wright EM . Biological membranes: the physical basis of ion and nonelectrolyte selectivity. Annu Rev Physiol 1969;31:581–646.
Claude P . Morphological factors influencing transepithelial permeability: a model for the resistance of the zonula occludens. J Membr Biol 1978;39:219–232.
Furuse M, Fujita K, Hiiragi T, et al. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 1998;141:1539–1550.
Furuse M, Hirase T, Itoh M, et al. Occludin: A novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–1788.
Cordenonsi M, D'Atri F, Hammar E, et al. Cingulin contains globular and coiled-coil domains and interacts with ZO-1, ZO-2, ZO-3, and myosin. J Cell Biol 1999;147:1569–1582.
Haskins J, Gu L, Wittchen ES, et al. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol 1998;141:199–208.
Citi S, Sabanay H, Jakes R, et al. Cingulin, a new peripheral component of tight junctions. Nature 1988;333:272–275.
Jesaitis LA, Goodenough DA . Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J Cell Biol 1994;124:949–961.
Stevenson BR, Siliciano JD, Mooseker MS, et al. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (Zonula Occludens) in a variety of epithelia. J Cell Biol 1986;103:755–766.
Van Itallie C, Rahner C, Anderson JM . Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J Clin Invest 2001;107:1319–1327.
Colegio OR, Van Itallie C, Rahner C, et al. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am J Physiol Cell Physiol 2003;284:C1346–C1354.
Van Itallie CM, Fanning AS, Anderson JM . Reversal of charge selectivity in cation or anion-selective epithelial lines by expression of different claudins. Am J Physiol Renal Physiol 2003;285:F1078–F1084.
Simon DB, Lu Y, Choate KA, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 1999;285:103–106.
Gitelman HJ, Graham JB, Welt LG . A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians 1966;79:221–235.
Ma TY, Hoa NT, Tran DD, et al. Cytochalasin B modulation of Caco-2 tight junction barrier: role of myosin light chain kinase. Am J Physiol Gastrointest Liver Physiol 2000;279:G875–G885.
Fanning AS, Jameson BJ, Jesaitis LA, et al. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem 1998;273:29745–29753.
Turner JR . ‘Putting the squeeze’ on the tight junction: understanding cytoskeletal regulation. Semin Cell Dev Biol 2000;11:301–308.
Meddings JB, Westergaard H . Intestinal glucose transport using perfused rat jejunum in vivo: model analysis and derivation of corrected kinetic constants. Clin Sci (Lond) 1989;76:403–413.
Pappenheimer JR . On the coupling of membrane digestion with intestinal absorption of sugars and amino acids. Am J Physiol 1993;265:G409–G417.
Turner JR, Cohen DE, Mrsny RJ, et al. Noninvasive in vivo analysis of human small intestinal paracellular absorption: regulation by Na+–glucose cotransport. Dig Dis Sci 2000;45:2122–2126.
Pappenheimer JR . Physiological regulation of transepithelial impedance in the intestinal mucosa of rats and hamsters. J Membr Biol 1987;100:137–148.
Turner JR, Rill BK, Carlson SL, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol 1997;273:C1378–C1385.
Berglund JJ, Riegler M, Zolotarevsky Y, et al. Regulation of human jejunal transmucosal resistance and MLC phosphorylation by Na(+)–glucose cotransport. Am J Physiol Gastrointest Liver Physiol 2001;281:G1487–G1493.
Turner JR, Black ED, Ward J, et al. Transepithelial resistance can be regulated by the intestinal brush-border Na(+)/H(+) exchanger NHE3. Am J Physiol Cell Physiol 2000;279:C1918–C1924.
Turner JR . Show me the pathway! Regulation of paracellular permeability by Na(+)–glucose cotransport. Adv Drug Deliv Rev 2000;41:265–281.
Nusrat A, Turner JR, Madara JL . Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am J Physiol Gastrointest Liver Physiol 2000;279:G851–G857.
Wainwright MS, Rossi J, Schavocky J, et al. Protein kinase involved in lung injury susceptibility: evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc Natl Acad Sci USA 2003;100:6233–6238.
Yuhan R, Koutsouris A, Savkovic SD, et al. Enteropathogenic Escherichia coli-induced myosin light chain phosphorylation alters intestinal epithelial permeability. Gastroenterology 1997;113:1873–1882.
MacDonald TT, Hutchings P, Choy MY, et al. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol 1990;81:301–305.
Fais S, Capobianchi MR, Silvestri M, et al. Interferon expression in Crohn's disease patients: increased interferon-gamma and -alpha mRNA in the intestinal lamina propria mononuclear cells. J Interferon Res 1994;14:235–238.
Mullin JM, Laughlin KV, Marano CW, et al. Modulation of tumor necrosis factor-induced increase in renal (LLC-PK1) transepithelial permeability. Am J Physiol 1992;263:F915–F924.
Madara JL, Stafford J . Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest 1989;83:724–727.
Taylor CT, Dzus AL, Colgan SP . Autocrine regulation of epithelial permeability by hypoxia: role for polarized release of tumor necrosis factor alpha. Gastroenterology 1998;114:657–668.
Hoffmann JC, Pawlowski NN, Kuhl AA, et al. Animal models of inflammatory bowel disease: an overview. Pathobiology 2002;70:121–130.
Cooke KR, Hill GR, Crawford JM, et al. Tumor necrosis factor- alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest 1998;102:1882–1891.
Cooke KR, Gerbitz A, Crawford JM, et al. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J Clin Invest 2001;107:1581–1589.
Davidson NJ, Leach MW, Fort MM, et al. T helper cell 1-type CD4+ T cells, but not B cells, mediate colitis in interleukin 10-deficient mice. J Exp Med 1996;184:241–251.
Madsen KL, Malfair D, Gray D, et al. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis 1999;5:262–270.
Bristol IJ, Farmer MA, Cong Y, et al. Heritable susceptibility for colitis in mice induced by IL-10 deficiency. Inflamm Bowel Dis 2000;6:290–302.
Hermiston ML, Gordon JI . Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science 1995;270:1203–1207.
Baert FJ, D'Haens GR, Peeters M, et al. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn's ileocolitis. Gastroenterology 1999;116:22–28.
Van den Brande JM, Braat H, van den Brink GR, et al. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn's disease. Gastroenterology 2003;124:1774–1785.
Agnholt J, Kaltoft K . Infliximab downregulates interferon-gamma production in activated gut T-lymphocytes from patients with Crohn's disease. Cytokine 2001;15:212–222.
Suenaert P, Bulteel V, Lemmens L, et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol 2002;97:2000–2004.
Brown GR, Lindberg G, Meddings J, et al. Tumor necrosis factor inhibitor ameliorates murine intestinal graft-versus-host disease. Gastroenterology 1999;116:593–601.
Schreiber S, Fedorak RN, Nielsen OH, et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn's disease. Crohn's Disease IL-10 Cooperative Study Group. Gastroenterology 2000;119:1461–1472.
Fedorak RN, Gangl A, Elson CO, et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn's disease. The Interleukin 10 Inflammatory Bowel Disease Cooperative Study Group. Gastroenterology 2000;119:1473–1482.
Madsen KL, Lewis SA, Tavernini MM, et al. Interleukin 10 prevents cytokine-induced disruption of T84 monolayer barrier integrity and limits chloride secretion. Gastroenterology 1997;113:151–159.
Ferrier L, Mazelin L, Cenac N, et al. Stress-induced disruption of colonic epithelial barrier: role of interferon-gamma and myosin light chain kinase in mice. Gastroenterology 2003;125:795–804.
Jenkins RT, Jones DB, Goodacre RL, et al. Reversibility of increased intestinal permeability to 51Cr-EDTA in patients with gastrointestinal inflammatory diseases. Am J Gastroenterol 1987;82:1159–1164.
Woywodt A, Ludwig D, Neustock P, et al. Mucosal cytokine expression, cellular markers and adhesion molecules in inflammatory bowel disease. Eur J Gastroenterol Hepatol 1999;11:267–276.
Nejdfors P, Wang Q, Ekelund M, et al. Increased colonic permeability in patients with ulcerative colitis: an in vitro study. Scand J Gastroenterol 1998;33:749–753.
Su C, Salzberg BA, Lewis JD, et al. Efficacy of anti-tumor necrosis factor therapy in patients with ulcerative colitis. Am J Gastroenterol 2002;97:2577–2584.
Papadakis KA, Treyzon L, Abreu MT, et al. Infliximab in the treatment of medically refractory indeterminate colitis. Aliment Pharmacol Ther 2003;18:741–747.
Scheinin T, Butler DM, Salway F, et al. Validation of the interleukin-10 knockout mouse model of colitis: antitumour necrosis factor-antibodies suppress the progression of colitis. Clin Exp Immunol 2003;133:38–43.
Musch MW, Clarke LL, Mamah D, et al. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting epithelial Na+/K+-ATPase. J Clin Invest 2002;110:1739–1747.
Radojevic N, McKay DM, Merger M, et al. Characterization of enteric functional changes evoked by in vivo anti-CD3 T cell activation. Am J Physiol 1999;276:R715–R723.
Fegan C, Poynton CH, Whittaker JA . The gut mucosal barrier in bone marrow transplantation. Bone Marrow Transplant 1990;5:373–377.
Robinet E, Ibrahim A, Truneh A, et al. Serum levels and receptor expression of tumor necrosis factor-alpha following human allogeneic and autologous bone marrow transplantation. Transplantation 1992;53:574–579.
Jacobsohn DA, Hallick J, Anders V, et al. Infliximab for steroid-refractory acute GVHD: a case series. Am J Hematol 2003;74:119–124.
Kobbe G, Schneider P, Rohr U, et al. Treatment of severe steroid refractory acute graft-versus-host disease with infliximab, a chimeric human/mouse antiTNFalpha antibody. Bone Marrow Transplant 2001;28:47–49.
Simpson D . New developments in the prophylaxis and treatment of graft versus host disease. Expert Opin Pharmacother 2001;2:1109–1117.
Lahat N, Shapiro S, Karban A, et al. Cytokine profile in coeliac disease. Scand J Immunol 1999;49:441–446.
Gillett HR, Arnott ID, McIntyre M, et al. Successful infliximab treatment for steroid-refractory celiac disease: a case report. Gastroenterology 2002;122:800–805.
Riegler M, Sedivy R, Pothoulakis C, et al. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Invest 1995;95:2004–2011.
Moore R, Pothoulakis C, LaMont JT, et al. C. difficile toxin A increases intestinal permeability and induces Cl- secretion. Am J Physiol 1990;259:G165–G172.
Triadafilopoulos G, Pothoulakis C, Weiss R, et al. Comparative study of Clostridium difficile toxin A and cholera toxin in rabbit ileum. Gastroenterology 1989;97:1186–1192.
Acknowledgements
We appreciate the comments of Drs. S. Blink, Y.-X. Fu, and S. Kane. D.R.C. is a predoctoral fellow of the National Institutes of Health (The University of Chicago Medical Scientist Training Program T32 GM07281). Work in Dr. Turner's laboratory was supported by the National Institutes of Health (R01DK61931), the Crohn's Colitis Foundation of America, the University of Chicago Digestive Disease Center (P30 DK42086), and the University of Chicago Cancer Center (P30 CA14599).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Clayburgh, D., Shen, L. & Turner, J. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest 84, 282–291 (2004). https://doi.org/10.1038/labinvest.3700050
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700050
Keywords
This article is cited by
-
Microbiota-dependent presence of murine enteric glial cells requires myeloid differentiation primary response protein 88 signaling
Journal of Biosciences (2023)
-
ADT-OH improves intestinal barrier function and remodels the gut microbiota in DSS-induced colitis
Frontiers of Medicine (2023)
-
A bridge for short-chain fatty acids to affect inflammatory bowel disease, type 1 diabetes, and non-alcoholic fatty liver disease positively: by changing gut barrier
European Journal of Nutrition (2021)
-
3D-Printed electrochemical sensor-integrated transwell systems
Microsystems & Nanoengineering (2020)
-
Using high-throughput sequencing to explore the anti-inflammatory effects of α-mangostin
Scientific Reports (2019)
