
Abstract
Hypertension and brachydactyly syndrome (HTNB) with short stature is an autosomal-dominant disorder. Mutations in the PDE3A gene located at 12p12.2-p11.2 were recently identified in HTNB families. We found a novel heterozygous missense mutation c.1336T>C in exon 4 of the PDE3A gene in a Japanese family with multiple HTNB patients. This mutation was found to be completely linked to the family members who inherited this condition. The mutation, resulting in p.Ser446Pro, was located within the cluster region of reported mutations. This mutation may also affect the phosphodiesterase activity of PDE3A to reduce the cyclic AMP level in the cell and thereby influencing the development of limbs and the function of the cardiovascular system.
Similar content being viewed by others
Introduction
Hypertension and brachydactyly syndrome (HTNB; MIM 112410) is characterized by a brachydactyly type E (BDE) with salt-independent and age-dependent hypertension.1 Affected patients manifest severe hypertension from childhood. The first report of this disease described a large Turkish family with brachydactyly, short stature and hypertension that was exacerbated with age and, if untreated, led to the onset of stroke before 50 years of age.2 Individuals showing BDE with shorter stature (~10 cm) tend to have a lower mean birth weight, stocky build and a rounder face than unaffected siblings, although the phenotype varies within an affected family.1 A linkage study of the aforementioned Turkish family identified the location of the candidate genes at 12p12.2-p11.2.3
A susceptible locus for essential hypertension had also been identified on chromosome 12p through genome-wide linkage analysis of a large Chinese kindred and additional nuclear families.4 This locus overlaps that identified in the Turkish family. Additionally, several reports such as a de novo deletion at 12p11.21-p12.2 from a Japanese patient manifesting brachydactyly with borderline high blood pressure for age,5 and complex rearrangements of the 12p11.2-p12.1 region in a patient with hypertension and brachydactyly,6 have strongly suggested that the causative genes are in this region, although the molecular mechanisms underlying this phenotype remain to be elucidated.2 Recently, Maass et al.7 identified mutations in the PDE3A gene at 12p12.2 from six families with HTNB. These mutations upregulated the cAMP hydrolytic activity that produces lower cAMP levels in the cell which may activate arteriogenesis leading to hypertension, in addition to the regulation of chondrogenesis by decreasing the expression of PTHLH and ultimately causing brachydactyly.
We here report a mutation in the PDE3A gene in a Japanese family with HTNB. This mutation was located within the cluster of mutations reported previously.7 From our analysis, we postulate a similar mechanism to that proposed previously in which hyperactivate cAMP hydrolytic activity resulted in BDE with hypertension in our patient.
Materials and methods
Subjects
After approval by the Ethical Review Board for Human Genome Studies at Fujita Health University and written informed consent, we obtained blood or saliva samples from the patient and his family members. High-molecular genomic DNAs were extracted from the blood or saliva using QuickGene 610L (Fuji film, Tokyo, Japan) or the Oragene Kit (DNA Genotek, Kanata, Ontario, Canada), respectively.
Cytogenetic microarray
About 50 ng of the study patient’s DNA was used for analysis with a Cytoscan HD Array (Affymetrix, Santa Clara, CA, USA) in accordance with the manufacturer’s instructions. Regions of copy-number changes were extracted with 20 probes of 50 kb using Chromosome Analysis Suite 3.0 (Affymetrix). All of the extracted regions containing a copy-number change were checked by visual comparisons with the normal control data from Database of Genomic Variants (http://dgv.tcag.ca).8
Exome sequencing
Fifty nanograms of patient DNA was used to generate a whole-exome library using SureSelect QXT Reagent and SureSelect Clinical Research Exome (Agilent Technologies, Santa Clara, CA, USA). Sequencing was carried out using HiSeq 1500 (Illumina, San Diego, CA, USA). After demultiplexing from other sample data, the reads were mapped onto the human reference hg19 using BWA 0.7.10.9 Sorting and recalibration of the mapped reads were carried out using Picard tools 1.118 (http://picard.sourceforge.net) and samtools 0.1.19,10 and variants were called into a VCF file using GATK 3.3–0.11 Annotations were added using Variant Studio 2.3 (Illumina). Low-quality data and common single-nucleotide polymorphisms (SNPs) of >5% frequency in Asian or global populations were filtered out. In our search of the candidate region at 12p12.2-p11.2, we identified one heterozygous missense mutation in the PDE3A gene. The mutation was confirmed by Sanger sequencing of PCR-amplified products for the corresponding region using the primer pair 5′-CTCTTTCCTAGCGCCTGAG-3′ and 5′-TGGATGAAGGTGCTTCCTG-3′ and amplification conditions of 94 °C for 2 min, 30 cycles of 30 s at 94 °C and 30 s at 63 °C, followed by 5 min at 63 °C using 20 ng of DNA and ExTaq (TaKaRa BIO, Kyoto, Japan). Mutations in other family members were detected by direct sequencing of PCR products.
Prediction of possible impact of amino-acid substitutions on protein function
PolyPhen-2 (version 2.2.2, http://genetics.bwh.harvard.edu/pph2/) predicts damaging effects of missense mutations based on the similarity to the homolog of other species and the chemical features of the amino acids or the positional relationship to the protein structure or domains.12 SIFT (http://sift.jcvi.org) and PROVEAN (http://provean.jcvi.org/index.php) prediction is based on the weighting of evolutionarily conserved amino-acid positions across multiple species.13, 14
Results and Discussion
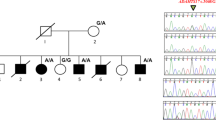
Figure 1a illustrates the pedigree of the patient’s family. The proband (III-1) was an 8-year-old boy of short stature (−1.9 s.d.), with short fingers and toes and normal-sized palms, and normal balanced bones (Figure 1b). He was diagnosed with myocardial hypertrophy with a systolic and diastolic blood pressure of 120 and 82 mm Hg, respectively. There was no appearance of obesity or mental retardation. His father (II-1) was also of short stature (150 cm) with short fingers and toes and had been taking medications for hypertension since the age of 23 years (a systolic blood pressure of 200 mm Hg). The paternal grandmother (I-2) was also of short stature with short fingers and toes and had hypertension from young adulthood. The patient’s brother (III-2) had been recently born and had similarly characteristic short fingers and toes (Figure 1c).

Characteristics of the study patient with HTNB and his family. (a) Pedigree of the family. (b) Photo and radiographic image of the patient’s hands with short fingers. (c) Photo of the hands of the patient’s brother (III-2). A full color version of this figure is available at the Journal of Human Genetics journal online.
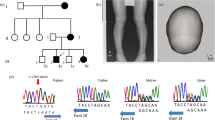
We first performed cytogenetic microarray analysis to detect copy-number mutations in our patient, but all of the identified copy-number variations were confirmed to be benign (data not shown). We then searched for nucleotide changes in the candidate region, 12p12.2-p11.2, by whole-exome analysis of the patient. We screened the regions encompassing the PDE3A and BICD1 genes as the precise location of the deletion in a HTNB patient had been reported previously.15 Among the 80 annotated genes, we found one missense change, c.1336T>C (p.Ser446Pro), in exon 4 of PDE3A (Figure 2a). No other changes were found in the exons of other genes, including PTHLH.

The c.1336T>C mutation in the patient’s family. (a) Sanger sequencing of the mutation hot spot region7 in the PDE3A gene in healthy controls. The mutation resulted in a p.Ser446Pro change at the hot spot region. (b) Sanger sequencing of the healthy grandfather (I-1), affected grandmother (I-2), affected father (II-1), healthy mother (II-2), patient (III-1) and affected brother (III-2). A full color version of this figure is available at the Journal of Human Genetics journal online.
There has been no report of the c.1336T>C (p.Ser446Pro) change as an SNP in dbSNP141 (URL: http://www.ncbi.nlm.nih.gov/projects/SNP/index.html) nor in the Japanese database HGVB (URL: http://www.genome.med.kyoto-u.ac.jp/SnpDB).16 By Sanger sequencing, this mutation was revealed to be inherited from the patient’s father who also had HTNB. The grandmother and the brother of the patient also had the same mutation and similar phenotype, indicating a tight link to the phenotype (Figure 2b).
Recently, Maass et al.7 described five PDE3A mutations in six families with HTNB. All were missense mutations within a cluster of five amino acids in exon 4 (Figure 2a) that is almost completely conserved among vertebrates.7 In that study also, HeLa cells expressing the mutant PDE3A exhibited reduced cAMP levels compared with those expressing the wild-type protein, indicating that this is a gain-of-function mutation that causes an increase in the cAMP-hydrolytic activity of the protein. The authors concluded that the region containing the mutation may enhance the phosphorylation of the PDE3A protein and induce a hyperactive state. The mutation in our current patient’s family was also found within the five amino-acid cluster (Figures 2a and b), suggesting that PDE3A hyperactivation is part of the mechanism leading to HTNB.
We calculated the prediction of possible impact of amino-acid substitutions on protein function. PolyPhen-2,12 SIFT13 and PROVEAN14 prediction all showed higher scores, indicating damaging or deleterious to the protein of the p.Ser446Pro substitution (Supplementary Table S1). As similar results were obtained for other PDE3A substitutions reported by Maass et al.7 (Supplementary Table S1), p.Ser446Pro may also alter the protein characteristics, possibly through the hyperactivation when phosphorylated.
The mutations associated with HTNB produce a specific enhancement of cAMP degradation but do not cause abnormal levels of cGMP. This increases the chance of developing medications as cAMP degradation activity can be specifically targeted. In this regard, milrinone is a specific inhibitor of PDE3A. However, Maass et al.7 have reported that high concentrations of this drug are necessary to inhibit the activity of certain types of mutant PDE3A as compared with the wild-type protein. New types of drugs are thus likely to be needed to prevent stroke in patients with HTNB.
References
Pereda, A., Garin, I., Garcia-Barcina, M., Gener, B., Beristain, E., Ibañez, A. E. et al. Brachydactyly E: isolated or as a feature of a syndrome. Orphanet J. Rare Dis. 8, 141 (2013).
Bilginturan, N., Zileli, S., Karacadag, S. & Pirnar, T. Hereditary brachydactyly associated with hypertension. J. Med. Genet. 10, 253–259 (1973).
Schuster, H., Wienker, T. F., Bähring, S., Bilginturan, N., Toka, H. R., Neitzel, H. et al. Severe autosomal dominant hypertension and brachydactyly in a unique Turkish kindred maps to human chromosome 12. Nat. Genet. 13, 98–100 (1996).
Gong, M., Zhang, H., Schulz, H., Lee, Y. A., Sun, K., Bähring, S. et al. Genome-wide linkage reveals a locus for human essential (primary) hypertension on chromosome 12p. Hum. Mol. Genet. 12, 1273–1277 (2003).
Bähring, S., Nagai, T., Toka, H. R., Nitz, I., Toka, O., Aydin, A. et al. Deletion at 12p in a Japanese child with brachydactyly overlaps the assigned locus of brachydactyly with hypertension in a Turkish family. Am. J. Hum. Genet. 60, 732–735 (1997).
Bähring, S., Rauch, A., Toka, O., Schroeder, C., Hesse, C., Siedler, H. et al. Autosomal-dominant hypertension with type E brachydactyly is caused by rearrangement on the short arm of chromosome 12. Hypertension 43, 471–476 (2004).
Maass, P. G., Aydin, A., Luft, F. C., Schächterle, C., Weise, A., Stricker, S. et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 47, 647–653 (2015).
MacDonald, J. R., Ziman, R., Yuen, R. K., Feuk, L. & Scherer, S. W. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 12, D986–D992 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N. et al. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7, e46688 (2012).
Lu, H. Y., Cui, Y. X., Shi, Y. C., Xia, X. Y., Liang, Q., Yao, B. et al. A girl with distinctive features of borderline high blood pressure, short stature, characteristic brachydactyly, and 11.47Mb deletion in 12p11.21-12p12.2 by oligonucleotide array CGH. Am. J. Med. Genet. A 149A, 2321–2323 (2009).
Narahara, M., Higasa, K., Nakamura, S., Tabara, Y., Kawaguchi, T., Ishii, M. et al. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS ONE 9, e100924 (2014).
Acknowledgements
We thank the patients and their families for participating in this study. We also thank to E Hosoba for technical assistance and T Ohye, M Tsutsumi and T Kato for helpful discussions. This work was support by the MEXT-Supported Program for the Strategic Research Foundation at Private Universities from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Boda, H., Uchida, H., Takaiso, N. et al. A PDE3A mutation in familial hypertension and brachydactyly syndrome. J Hum Genet 61, 701–703 (2016). https://doi.org/10.1038/jhg.2016.32
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.32
This article is cited by
-
An aggressive systemic mastocytosis preceded by ovarian dysgerminoma
BMC Cancer (2020)
-
Whole-exome sequencing identifies a de novo PDE3A variant causing autosomal dominant hypertension with brachydactyly type E syndrome: a case report
BMC Medical Genetics (2020)
-
A genome-wide association study for harness racing success in the Norwegian-Swedish coldblooded trotter reveals genes for learning and energy metabolism
BMC Genetics (2018)
-
RNA Sequencing Reveals Novel Transcripts from Sympathetic Stellate Ganglia During Cardiac Sympathetic Hyperactivity
Scientific Reports (2018)
-
Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement
Nature Reviews Endocrinology (2018)
