
Abstract
In a patient suspected clinically to have Weaver syndrome, we ruled out mutations in EZH2 and NSD1, then identified a previously undescribed de novo mutation in EZH2’s partner protein EED. Both proteins are members of the Polycomb Repressive Complex 2 that maintains gene silencing. On the basis of the similarities of the patient’s phenotype to Weaver syndrome, which is caused by de novo mutations in EZH2, and on other lines of evidence including mouse Eed hypomorphs, we characterize this mutation as probably pathogenic for a Weaver-like overgrowth syndrome. This is the first report of overgrowth and related phenotypes associated with a constitutional mutation in human EED.
Similar content being viewed by others

Introduction
Overgrowth syndromes manifest increased mass of multiple tissues and often include advanced bone age, facial dysmorphism and intellectual disability. Mutations in epigenetic regulators frequently affect many aspects of growth and metabolism, causing either short or tall stature. ‘Classical’ overgrowth syndromes include Sotos (MIM #117550) and Weaver (MIM #277590), caused by germline mutations in NSD11 and EZH22, 3 respectively. Recently, de novo mutations in DNMT3A and SETD2, two other epigenetic regulators, have been shown to cause distinct overgrowth syndromes.4, 5 Somatic mutations in all of these genes have also been implicated in various cancers, particularly hematological malignancies. On the basis of this evidence, we hypothesized that rare de novo mutations in other epigenetic regulators, particularly other members of the Polycomb Repressive Complex (PRC) 2, might explain unsolved overgrowth cases. Here we report on a Turkish man with overgrowth, facial dysmorphism and intellectual disability, and identify a probably pathogenic mutation in EED, another PRC2 member.
Case report
Our proband was born full term to non-consanguineous Turkish parents. Birth weight was 4100 g (standard deviation score (SDS) +1.09) and birth length 52 cm (SDS +0.38). Birth head circumference was not recorded. Dysmorphic features in childhood included macrocephaly, large bifrontal diameter, ocular hypertelorism and large ears (Figures 1a and b). Mild intellectual disability (IQ: 60) with speech delay (first words at age 2 years, first sentences at age 5 years) and poor fine motor skills were present. Brain magnetic resonance imaging was normal. He developed epilepsy, with his first seizure occurring at 4.5 years. Electroencephalogram showed a generalized irregular wave pattern, and phenytoin and primidone reduced seizure frequency to 1–2 per year (with no seizures from age 27 years onwards). He also developed moderate myopia (prescription lenses −5.5 and −6.5 diopters). Echocardiogram found mild mitral regurgitation. He developed tall stature in childhood: height was 87 cm (SDS +4.2) at 13 months, 138 cm (SDS +5.1) at 5 years 7 months and 158 cm (SDS +4.6) at 8 years 8 months (WHO growth charts). Blood hormone levels including thyroid-stimulating hormone (1.4 IU ml−1), T4 (1.22 ng ml−1), insulin-like growth factor 1 (201 ng ml−1) and fasting glucose (98 mg dl−1 or 5.4 mmol l−1) were all normal. He also had an umbilical hernia, and required surgical correction for cryptorchidism and a post-traumatic patellar dislocation at age 9 years. Following his knee surgery, circulatory failure led to amputation of his right leg over the knee at age 11 years. X-rays at age 4 years revealed dense physeal bands within the proximal femurs, and at 14.5 years bone age was consistent with 15 years. Additional X-rays at various ages (data not shown) revealed significant scoliosis, abnormal flaring of the distal clavicles, distal ribs, and metaphyses of the distal radius, distal ulna, distal femur and proximal tibia. The metaphyses were also abnormally lucent. He had flattened glenoid fossae and humeral heads, as well as a flattened left acetabulum and femoral head. At the most recent examination at age 27 years, his final height was 190 cm (SDS +1.85) and head circumference 59 cm (SDS +1.46). He has since undergone bilateral cataract surgery (age 30 years 10 months). Photographs of the patient confirm dysmorphic features including hypertelorism (adult interpupillary distance 8 cm), downslanting palpebral fissures and retrognathia with a prominent crease between the lower lip and the chin (Figures 1a to 1h, Supplementary Table 1). He also has kyphoscoliosis, widely spaced nipples and several pigmented nevi (Figures 1e and f), as well as large hands with camptodactyly (Figure 1h). To date, he has had no malignant or premalignant phenotypes and his blood cell counts remain normal (red cells 4.9 × 1012 per liter, white cells 6.5 × 109 per liter and platelets 250 × 109 per liter). He has a younger sister who does not have any overgrowth or dysmorphic features.

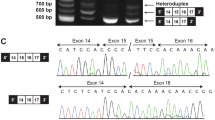
Characterization of patient with EED mutation. (a–h) Photographs of the proband show the features described in the paper at various ages: 3 years (a, b), 14 years (c, d) and 30 years (e–g); proband’s right hand is shown at 27 years of age (h). (i) Pedigree with Sanger confirmation that the patient carries a de novo c.1372A>C (p.R302S) mutation in EED. This mutation is absent in the sister who carries a variant in the adjacent residue; this variant is synonymous (p.T301=) and thus consistent with her lack of overgrowth and dysmorphic features. Sanger traces were analyzed using Sequence Scanner v1.0 (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA). A full color version of this figure is available at the Journal of Human Genetics journal online.
Whole-exome sequencing and results
Parents gave informed consent for participation in our study. EZH2 and NSD1 screening in the proband by Sanger sequencing were negative, so we constructed an Agilent (Santa Clara, CA, USA) All Exon V5+UTR capture library from his DNA before exome sequencing using Illumina (San Diego, CA, USA) HiSeq 2500. Read alignment and variant calling, filtering and annotation were carried out using our in-house pipeline. Variants unique to our patient (defined as not previously observed in public repositories or the in-house human variation database6) were prioritized. Variants consistent with the phenotype were analyzed further; top candidates were validated by Sanger sequencing in the quartet.
We identified a novel c.1372A>C missense mutation in EED (NM_003797.3), confirmed as de novo by Sanger sequencing (Figure 1i), present only in the proband. This variant is predicted to convert arginine residue 302 to serine, scored as damaging by Polyphen (http://genetics.bwh.harvard.edu/pph2/) although not by SIFT (http://sift.jcvi.org/) (Table 1). We also found and Sanger-validated two novel variants in MEGF8 (NM_001410.2): c.4517C>A inherited from the mother and c.3567G>A from the father, both absent in the sister (Supplementary Figure 1). Although this gene has been associated with an autosomal recessive subtype of Carpenter syndrome (MIM #614976),7 the proband had no major features of this syndrome (lacking polydactyly, heterotaxia or obvious craniosynostosis) and both variants were predicted to be benign (Table 1).
Discussion
We believe the de novo EED mutation is the most likely cause of the patient’s excessive height in childhood (max SDS +5.1). We found no other coding variants that could plausibly explain these features. Carpenter syndrome, suggested by the two rare variants in MEGF8,7 does feature some of the phenotypic traits observed in our patient (e.g. intellectual disability, cryptorchidism and camptodactyly). However, he had none of the cardinal manifestations of this latter syndrome (Supplementary Table 1).
EED is required for proper methyltransferase activity of EZH2.8 It is highly conserved in mammals with 100% amino-acid identity between mouse Eed and human EED, despite significant coding nucleotide differences between the Eed and EED genes.9 This form of conservation suggests that disruption of any given residue is likely to affect protein function. EED contains seven WD40 domains,10, 11 of which five appear functionally necessary12 with potentially specific roles.13 Our patient’s mutation (c.1372A>C, encoding p.Arg302Ser or R302S) falls within a conserved nucleotide region9 that affects all EED isoforms.12 It localizes within a WD40 domain that is required for H3K27 methylation12 and is possibly necessary for the EED–EZH2 interaction,12 which is in turn required for EZH2’s methyltransferase activity. Consistent with this hypothesis, mutations in one of the WD40 repeats in Eed block its interaction with Ezh2.14 The region surrounding residue 302 is thought to be involved in binding of EED to PRC2 and also to PRC1.15 This region appears to have a more important role in binding directly to H3K27, through a small pocket on the surface of EED formed by hydrophobic residues including C324, Y364 and nearby residue Y308.11, 16 Of interest, a mutation at Y358, six amino acids away from Y364 (the same distance as R302 to Y308), reduced EED binding to histone peptides by twofold.11
Further lines of evidence supporting the hypothesis that the R302S mutation disrupts EED function are summarized in Table 2. Hypothesis-testing experiments have shown that hypomorphic mutations in Eed cause developmental defects in mice. In addition, rare structural variants (deletions) including human EED co-occur with developmental defects that manifest in childhood. Rare somatic mutations in EED have also been observed in human cancers. In particular, p.R302G (a mutation affecting the same amino acid) has been reported in a myeloproliferative neoplasm17 (a type of tumor that has recurrent mutations in other overgrowth genes such as EZH2 and DNMT3A), reinforcing the concept that this particular residue is likely important for normal EED function.
The fact that common amino-acid polymorphisms in EED have not been observed across multiple healthy controls strongly suggests that sequence variation at the protein level is not compatible with good health, at least at the whole organism level. Given the known effects on growth of coding mutations in EZH2, we posited a high prior probability that functional mutations in other members of the PRC2 complex, such as EED, would likely cause overgrowth. Having now observed overgrowth in a patient with a de novo coding mutation in EED, we conclude that this patient’s overgrowth is attributable to this mutation.
References
Kurotaki, N., Imaizumi, K., Harada, N., Masuno, M., Kondoh, T., Nagai, T. et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 30, 365–366 (2002).
Gibson, W. T., Hood, R. L., Zhan, S. H., Bulman, D. E., Fejes, A. P., Moore, R. et al. Mutations in EZH2 cause Weaver syndrome. Am. J. Hum. Genet. 90, 110–118 (2012).
Tatton-Brown, K., Murray, A., Hanks, S., Douglas, J., Armstrong, R., Banka, S. et al. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am. J. Med. Genet. A 161A, 2972–2980 (2013).
Tatton-Brown, K., Seal, S., Ruark, E., Harmer, J., Ramsay, E., Del Vecchio Duarte, S. et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 46, 385–388 (2014).
Luscan, A., Laurendeau, I., Malan, V., Francannet, C., Odent, S., Giuliano, F. et al. Mutations in SETD2 cause a novel overgrowth condition. J. Med. Genet. 51, 512–517 (2014).
Fejes, A. P., Khodabakhshi, A. H., Birol, I. & Jones, S. J. Human variation database: an open-source database template for genomic discovery. Bioinformatics 27, 1155–1156 (2011).
Twigg, S. R., Lloyd, D., Jenkins, D., Elçioglu, N. E., Cooper, C. D., Al-Sannaa, N. et al. Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am. J. Hum. Genet. 91, 897–905 (2012).
Cao, R. & Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 15, 57–67 (2004).
Schumacher, A., Lichtarge, O., Schwartz, S. & Magnuson, T. The murine Polycomb-group gene eed and its human orthologue: functional implications of evolutionary conservation. Genomics 54, 79–88 (1998).
Han, Z., Xing, X., Hu, M., Zhang, Y., Liu, P. & Chai, J. Structural basis of EZH2 recognition by EED. Structure 15, 1306–1315 (2007).
Margueron, R., Justin, N., Ohno, K., Sharpe, M. L., Son, J., Drury, W. J. 3rd et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767 (2009).
Montgomery, N. D., Yee, D., Montgomery, S. A. & Magnuson, T. Molecular and functional mapping of EED motifs required for PRC2-dependent histone methylation. J. Mol. Biol. 374, 1145–1157 (2007).
Sathe, S. S. & Harte, P. J. The Drosophila extra sex combs protein contains WD motifs essential for its function as a repressor of homeotic genes. Mech. Dev. 52, 77–87 (1995).
Denisenko, O., Shnyreva, M., Suzuki, H. & Bomsztyk, K. Point mutations in the WD40 domain of Eed block its interaction with Ezh2. Mol. Cell. Biol. 18, 5634–5642 (1998).
Cao, Q., Wang, X., Zhao, M., Yang, R., Malik, R. & Qiao, Y. et al. The central role of EED in the orchestration of polycomb group complexes. Nat. Commun. 5, 3127 (2014).
Migliori, V., Mapelli, M. & Guccione, E. On WD40 proteins: propelling our knowledge of transcriptional control? Epigenetics 7, 815–822 (2012).
Nangalia, J., Massie, C. E., Baxter, E. J., Nice, F. L., Gundem, G., Wedge, D. C. et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. New Engl. J. Med. 369, 2391–2405 (2013).
Kaminsky, E. B., Kaul, V., Paschall, J., Church, D. M., Bunke, B., Kunig, D. et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet. Med. 13, 777–784 (2011).
Shumacher, A., Faust, C. & Magnuson, T. Positional cloning of a global regulator of anterior-posterior patterning in mice. Nature 383, 250–253 (1996).
Ng, J., Hart, C. M., Morgan, K. & Simon, J. A. A Drosophila ESC-E(Z) protein complex is distinct from other polycomb group complexes and contains covalently modified ESC. Mol. Cell. Biol. 20, 3069–3078 (2000).
Acknowledgements
We are grateful to the staff of the Library Construction, Sequencing and Bioinformatics teams at the Michael Smith Genome Sciences Centre for the data generation. We gratefully acknowledge the assistance of Professor Dr Nurten Akarsu of the TÜBİTAK Advanced Genomics and Bioinformatics Research Group NGS infrastructure for querying their in-house database of 587 Turkish exomes. We also thank the Centre for Applied Neurogenetics (UBC) for confirming family relationships on our samples. This work was supported by the CIHR (Operating Grants PCN102990, PCN110794, MOP119595), and by Clinician Scientist salary awards to WTG (CIHR and Child and Family Research Institute). ASAC holds a Doctoral Grant from the Fundação para a Ciência e a Tecnologia (Portugal/EU).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Cohen, A., Tuysuz, B., Shen, Y. et al. A novel mutation in EED associated with overgrowth. J Hum Genet 60, 339–342 (2015). https://doi.org/10.1038/jhg.2015.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.26
This article is cited by
-
Correlation between large rearrangements and patient phenotypes in NF1 deletion syndrome: an update and review
BMC Medical Genomics (2024)
-
Regulation, functions and transmission of bivalent chromatin during mammalian development
Nature Reviews Molecular Cell Biology (2023)
-
Transient Polycomb activity represses developmental genes in growing oocytes
Clinical Epigenetics (2022)
-
Interplay between chromatin marks in development and disease
Nature Reviews Genetics (2022)
-
The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective
Cellular and Molecular Life Sciences (2021)
