
Abstract
Microbial community composition and diversity change along chemical gradients, leading to the expectation that microbial community information might provide new gradient characterizations. Here we examine stream bacteria composition and diversity along a strong chemical gradient in Central Appalachian streams. Coal mining in the region generates alkaline mine drainage (AlkMD), causing dramatic increases in conductivity, alkalinity, sulfate and metals sufficient to degrade stream macrobiota communities throughout the ecoregion. In this study, we examined the relationship between water and biofilm chemistry and biofilm bacteria taxonomic composition in streams where active and reclaimed surface coal mines occupied 0–96% of watershed surface area. We incubated wood veneers in each stream site for 4 months to develop biofilms on similar substrates. We sampled water chemistry at the time of deployment and collection, and after 1 month. Following incubation, we collected biofilms for microbial and chemical characterization. Microbial composition was determined by pyrosequencing 16S rRNA amplicons. Biofilm subsamples were analyzed by inductively coupled plasma mass spectrometry to determine metal concentrations. Our results show that microbial community composition differed significantly between AlkMD-exposed and AlkMD-unexposed sites, and that compositional dissimilarity increased with AlkMD loading. Diversity was not correlated with pH or extent of upstream mining, but instead correlated with biofilm concentrations of Cd, Mn, Zn and Ni. Within mined sites, the extent of upstream mining was negatively correlated with taxonomic richness. Despite major compositional shifts, functional capacity predicted with PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) correlated with mining in only 3 of 43 level-2 KEGG (Kyoto Encyclopedia of Genes and Genomes) Orthology groups.
Similar content being viewed by others


Introduction
Microbial diversity, abundance and composition change along environmental gradients. Understanding the nature of these changes may help us identify a set of environmental conditions characteristic to particular groups of taxa (Feris et al., 2009; Logares et al., 2012). Ultimately, these relationships could be used to develop microbial indicators (Feris et al., 2009) and help predict microbial community responses to environmental conditions (Paerl et al., 2003; Sims et al., 2013). Communities of other organisms including macroinvertebrates and periphyton are widely and routinely used as bioindicators of localized chemical and physical conditions (Barbour et al., 1999; Hering et al., 2003; Solimini et al., 2006). Microbial communities, with their great genetic diversity and now rapid identification process, also hold promise as bioindicators. There is reason to believe that microbial community indexes or indicator taxa could be developed, as a variety of studies demonstrate that diversity and composition shift considerably along gradients of pH (Fierer and Jackson, 2006; Fierer et al., 2007; Lauber et al., 2009; Lear et al., 2009; Rousk et al., 2010; Griffiths et al., 2011), trace metal concentration (Baker and Banfield, 2003; Feris et al., 2003; Giller et al., 2009; Lami et al., 2013), salinity (Lozupone and Knight, 2007; Auguet et al., 2010) and substrate carbon-to-nitrogen ratio (Bates et al., 2011).
Because microbial composition can be affected strongly by pH, salinity and metal concentrations, we speculated that exposure to alkaline mine drainage (AlkMD) would drive important shifts in stream microbial assemblages. AlkMD results from surface coal mining, the dominant form of land cover change in Central Appalachia (Townsend et al., 2009). Effluent is produced during rock weathering of surface coal mines that contain carbonate rock strata in addition to coal layers (Palmer et al., 2010; Bernhardt and Palmer , 2011). The carbonate matrix buffers sulfuric acid produced from weathered pyrite minerals, increasing base cation (Ca2+, Mg2+ and HCO3–) and SO42- concentrations in receiving waters (Rose and Cravotta, 1998; Kirby and Cravotta, 2005). AlkMD is thus characterized by increased alkalinity, ionic strength and pH and often has elevated metals that reflect parent geology (Lindberg et al., 2011; USEPA, 2011; Griffith et al., 2012). Recent regional analyses suggest that AlkMD generated from surface mines led to significant chemical and biological degradation of at least 22% of southern West Virginia rivers (Bernhardt et al., 2012).
AlkMD pollution offers an interesting contrast to the large body of literature exploring responses to acid mine drainage, as it increases salinity and trace metals but enhances alkalinity rather than reduces pH. In this study, we compared stream bacterial communities between mined and unmined catchments within the largest surface coal mine complex in Central Appalachia. We asked the following questions. (1) Does AlkMD significantly alter community composition and do these changes manifest themselves as changes in α-diversity? (2) What bacterial taxa are responsible for changes in community composition and can these taxa be used as indicators of AlkMD? (3) What insight regarding functional response can be gained by examining taxa lost and gained because of the influence of AlkMD?
We expected bacteria’s compositional response to AlkMD would mirror that observed for macroorganisms (Pond, 2010; Bernhardt et al., 2012). Specifically, compositional shifts would be due to both decreased α-diversity and decreased evenness across the AlkMD gradient. We hypothesized that many taxa found in the low solute waters of unmined sites would be absent or rare in sites downstream of surface mines and that taxa known to metabolize ions released from mining would increase downstream of mining. We anticipated that taxa with distinct metabolic repertoires could indicate sites exposed to AlkMD.
Materials and methods
Study site
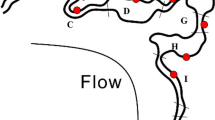
Sampling sites are in Mud River, a Central Appalachian surface coal mining region that lies in West Virginia’s Lower Guyandotte watershed (Figure 1). Mud River has two forks. Upper Mud River passes through the Hobet Mine complex, the largest surface coal mine in Central Appalachia and includes active and reclaimed mines within 40 km2 of permitted mines. Left Fork Mud River is unmined but has similar geology and low-density residential housing. Sampling locations spanned a gradient of AlkMD contamination. Sites affected by mining included 9 along Upper Mud River’s mainstem and 8 within tributaries draining mines (6 active and 2 reclaimed). Unmined sites were one in Upper Mud River upstream of surface mines, one unimpaired tributary and four Left Fork Mud River sites.

Sample sites on Mud River and Left Fork Mud River in Boone and Lincoln Counties, West Virginia (WV). Hydrologic unit codes (HUCs) 12-050701020301 and 12-050701020104 are outlined in gray. Gray tributary streams run through mined areas, whereas black tributaries are unmined. The mainstem of Mud River and Left Fork Mud River shown in bold black from headwaters to confluence. Arrows show flow direction. Inset of US mid-Atlantic states shows Appalachian Coalfield Region as gray-shaded area with relative location of study site in WV in red (not to scale).
Water chemistry
Water chemistry and temperature at each site were measured during deployment and collection of biofilm substrates and 1 month later (December 2010 and April and May 2011). We measured in-stream conductivity and pH and analyzed samples for a suite of major and trace elements (Supplementary Table S1). Water sampling followed USGS protocols (USGS). See Lindberg et al. (2011) for details.
Stream biofilms
Biofilms were grown on substrates suspended under water near the shaded streambank at each sampling site. To minimize variability (De Beer and Stoodley, 2006; Sabater et al., 2007) and use environmentally relevant substrates, we used wood veneers cut from the same tree (Acer saccharum) and enclosed veneers in mesh aquaculture bags (Pentair Aquatic Eco-Systems, Apopka, FL, USA). Four sterilized veneers were deployed under water at each site and incubated for 4 months. Veneers were removed in April 2011; two were transported to the lab on dry ice and stored at −80 °C until DNA extraction. Two remaining veneers were used for metal analysis and carbon content.
Biofilm scraped from two veneers was oven-dried at 78 °C, homogenized, digested with trace metal grade HNO3 and heated at 80 °C. Samples were analyzed for metal content with inductively coupled plasma mass spectrometry as detailed in Lindberg et al. (2011). Remaining biofilm was used for ash-free dry mass via combustion at 500 °C. Then, 53% of the difference between pre- and post- combusted dry mass was calculated as carbon content (Wetzel, 1983).
Biofilm community 16S rRNA gene analysis
See detailed methods in Supplementary Information. Briefly, we extracted DNA from homogenized biofilm using PowerBiofilm DNA isolation kit (MO BIO, Carlsbad, CA, USA) and amplified the 27–338 region of 16S ribosomal RNA (rRNA). Sequencing was unidirectional using Roche 454 Lib-L kit (Branford, CT, USA). Replicate PCR samples were pooled, purified and normalized before being sent to the Genome Sequencing and Analysis Core Resource at Duke University (Durham, NC, USA) for pyrosequencing with a Roche 454 Life Sciences Genome Sequencer Flex Titanium instrument.
Bacterial community analyses
QIIME 1.6.0 software pipeline (Caporaso et al., 2010) was used for downstream sequence processing: reverse primer and chimera removal, phylotype binning, operational taxonomic unit (OTU) assignment and 10-base MID (multiplex identifier) sample grouping. USEARCH was used to filter noisy sequences, chimera check and pick OTUs from demultiplexed sequences (Edgar, 2010). OTUs containing <3 sequences were removed. Remaining OTUs were picked at 97% sequence similarity and identified using the RDP (Ribosomal Database Project) classifier retrained with Greengenes. The NAST algorithm was used for alignment and Greengenes (http://greengenes.secondgenome.com) supplied core representative sequences (version October 2012). Sequences were quality filtered and rarefied to 1543 sequences per sample. To select 1543 sequences for analysis, each OTU’s abundance at a site was divided by the total sequence count for that site, and then multiplied by 1543 to retain the relative abundance of that OTU out of 1543 sequences. The resulting decimals were floored and remaining sequences needed for the site to contain 1543 sequences were selected using the distribution of OTUs at each site (Beevers, 2006). One unmined site (MRUl2) had only 825 sequences. Data from this site were used in environmental data correlations, but excluded from diversity calculations. Rarefaction curves were generated for Chao1 richness (Chao, 1984), Margalef’s index, Shannon diversity, Simpson’s index for evenness and evenness.
Multivariate analysis was guided by Anderson and Willis (2003), who advocate following these approaches: (1) an ordination (robust and unconstrained), (2) statistical testing of the hypothesis and (3) identification of taxa driving the observed patterns. We visualized differences in OTU-based community composition with nonmetric multi-dimensional scaling (NMDS) ordinations based on Bray–Curtis (Bray and Curtis, 1957) and generalized UniFrac (GUniFrac) distance matrices (item 1). GUniFrac distances measure community phylogenetic relatedness, but cover a series of distances from weighted to unweighted by adjusting the weight of the branches in the UPGMA tree (Chen et al., 2012). Alpha controls the weight on lineages with common taxa and was set to 0.5 to provide the best overall power (Chen et al., 2012). For analysis, we grouped mined sites in two different ways: a priori (mainstem mined, active valley fill and reclaimed valley fill) and post hoc, (Ward's method cluster analysis of Bray–Curtis distances separated sites into group A and group B) (Supplementary Figure S2). Finally, we partitioned variation in our community distance matrix among these a priori and post hoc groupings with permutational multivariate analysis of variance (item 3) (Anderson, 2001). See detailed methods in Supplementary Information.
Bacteria taxa and environmental analysis
To understand community composition and environmental variable associations, we examined correlations between NMDS ordination scores and the first two component scores derived from a principal components analysis (PCA) of transformed environmental variables. We used correlation to look for trends between relative abundance of all phyla and classes and PCA axes, pH gradient and percent of watershed area mined. Because examining linear relationships between taxonomic groups at high hierarchical levels can obscure taxa-specific patterns at lower levels, we also used generalized linear models (Quasi-Poisson regression; McCullagh and Nelder, 1989) to identify genera with positive (slope>0, P<0.10), negative (slope<0, P<0.10) and no response (P>0.1) across the gradient of area mined. Finally, we characterized taxa driving multivariate patterns using indicator species analysis (Dufrene and Legendre, 1997; De Cáceres and Legendre, 2009; De Cáceres et al., 2010) with PC-ORD software (McCune and Mefford, 2011) (Supplementary Information).
Predicted functional profiles
To predict functional responses to the mining gradient, we used PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States; http://picrust.github.com; Langille et al., 2013) to generate a functional profile using our 16S rRNA data. We followed the suggested methods for OTU picking with Greengenes 13.5 using Galaxy (http://huttenhower.sph.harvard.edu/galaxy/). Predicted gene family abundances were rarefied to 1078 sequences per site, analyzed at KEGG (Kyoto Encyclopedia of Genes and Genomes) Orthology group levels 2 and 3 and used in correlation analysis (Pearson’s) with percent watershed mined and the PCA axes. The mean nearest sequenced taxon index was lower (0.14±0.002) than that reported for soil communities (0.17±0.02) (Langille et al., 2013).
Results
Environmental characterization of streams
Our data set included sites that were unmined (n=5), Mud River tributaries draining active (n=6) and reclaimed (n=2) mines and sites within the mainstem of Mud River both upstream and downstream of mined tributaries (n=9). All mined sites had higher concentrations of typical AlkMD constituents (SO42−, Ca2+, Mg2+, Se and Mn) than unmined sites (Table 1 and Supplementary Figure S1), leading to a distinct chemical composition in a PCA (Figure 2). The majority of environmental variables strongly correlated with component 1 are classically associated with AlkMD, with SO42−, Ca2+ and Mg2+ all highly correlated with this first axis (Supplementary Table S3). Component 1 also strongly correlated with landcover such that the percentage of watershed mined that drained to a sampling location explained 96% of site variance (r=0.96, P <0.001). We also observed unexpected increases in non-purgeable organic carbon (r=0.79, P<0.001) and mean nitrate (r=0.72, P<0.001) along the component 1, or AlkMD, axis (Supplementary Table S3). Component 2 positively correlated with increasing biofilm Cd, Mn, Zn, Ni and Zn concentrations (r>0.6, P<0.01).

PCA of selected environmental variables. Component 1 and component 2 explain 56.8±3.8% and 16.9±2.1% variance, respectively. Sites with mining split into group A (heavily mined; symbols outlined) and group B (moderately mined; symbols not outlined).
Biofilm biomass as DNA per unit surface area ranged from 6 to 274 mg m−2 (mean=57±12.07 mg m−2) and did not differ between mined and unmined sites (log-transformed, Student’s t-test, P=0.25). Biofilm C content ranged from 10 to 127 g C m−2 (mean=42±6.17 g C m−2) and was not different between mined and unmined sites (log-transformed, Student’s t-test, P=0.24).
Sequencing and taxa identification
The sequencing run of 16S rRNA amplicons yielded 145 138 raw reads. Filtering and removing nontarget sites left 23 sites with 102 772 sequences. Maximum reads per site was 7555 with mean 3543 (s.e. 393). Final sequence clustering gave 1846 OTUs. Each sample had a mean of 391 OTUs (s.e. 9). Identification of OTUs at different taxonomic levels yielded 304 species, 298 genera, 203 families, 128 orders, 72 classes and 25 phyla. Raw sequences are available at MG-RAST (accession numbers 4498070.3–4498093.3, http://metagenomics.anl.gov/linkin.cgi?project=1572; Meyer et al., 2008).
Across all sites, Proteobacteria was the dominant phylum (66.7%), followed by Bacteroidetes (20.8%), Acidobacteria (4.7%) and Actinobacteria (2.0%). All other phyla had abundances of <1%. At the phylum level, <0.02% of reads were unclassified. The most common classes were Alphaproteobacteria (39.0%), Betaproteobacteria (19.3%) and Sphingobacteria (12.5%). At the class level, 2% of reads were unclassified. The most abundant genera were Flavobacterium (6.7%) and Novosphingobium (4.9%). Of the OTUs, 1.1% were assigned to known species.
Overall bacterial community structure
We determined compositional differences among the following site categories: unmined, within valley filled tributaries and in Mud River’s mainstem downstream of valley filled tributaries using Bray–Curtis distance and GUniFrac NMDS (Table 2). With the Bray–Curtis distance matrix, mined sites separated into two groups using hierarchical cluster analysis (Ward’s method), leading us to reclassify mined sites into two post hoc groupings: group A and group B (Figure 3a). Community composition of these two groups differed significantly (permutational multivariate analysis of variance, F2, 19=4.61, P<0.001, Table 2). Group A was characterized by higher concentrations of biofilm Ca, Cd, Mn, Ni, Sr, Th and Zn, and water column Ca, Ni, Se, SO42- and TN (Student’s t-test, P⩽0.05; Figure 4). Group A sites occurred in stream reaches draining watersheds with 25–96% of watershed area occupied by mines, whereas sites in group B had 16–51% of their watershed mined (Student’s t-test, P=0.03).

NMDS ordination (a) using Bray–Curtis distance matrix and (b) using GUniFrac distance matrix (with α=0.5). Mined sites categorized as group A (symbols outlined) and group B (symbols not outlined) resulting from cluster analysis of Bray–Curtis distance NMDS. Distance matrices based on 16S rRNA pyrosequences. The r2 values are in parentheses. Stress: (a) 0.192 and (b) 0.190. Rarefied to 1543 sequences per site.

Water and biofilm chemistry variables that differ significantly between groups A and B shown as proportion of change from average concentration in unmined sites (P⩽0.05).
Based on Bray–Curtis distances, which do not incorporate phylogenetic relatedness in community differences, bacteria community composition differed significantly overall (F3, 18 =1.94, P=0.002) and between sites with and without AlkMD (F3, 18=3.18, P<0.001; Figure 3a and Table 2). There were no significant differences in community composition between streams draining active and reclaimed valley fills (F1, 18=1.04, P=0.42). NMDS axes 1 and 2 had similar degrees of explanatory power (25.9% and 26.4%, respectively). Configuration stress was 0.192.
We also used GUniFrac to compare composition across sites. GUniFrac analyses include the phylogenetic relatedness of taxa. Results were similar to the Bray–Curtis analysis, although separation between sites in ordination space was less distinct (F3, 18=1.55, P=0.02; Figure 3b and Table 2). Contrasts between mined and unmined sites showed significant differences in bacterial community composition (F1, 18=2.36, P=0.002), but again we found no difference between communities in streams below reclaimed and active valley fills (F1, 18=0.72, P=0.75). Axis 1 in the GUniFrac distance NMDS ordination explained the most compositional differences between sites (31.0%), whereas axis 2 explained 21.3% and stress was 0.190.
Bacterial diversity along the mining gradient
We examined correlations between α-diversity and environmental variables. Component 2, a PCA axis capturing variation in biofilm metals, was the single strongest correlate of richness estimated by multiple α-diversity metrics. Chao1 richness, Margalef’s index and evenness negatively correlated with component 2 (Chao1: P=0.002, r=−0.63; Margalef’s index: P=0.006, r=−0.57; evenness: P=0.006, r=−0.57). Across all sites we did not observe significant correlations between percent of watershed mined and α-diversity metrics (Figure 5, all P>0.05). However, Chao1 richness estimator and Margalef’s index of only mined sites were significantly negatively correlated with the percent watershed mined (both P=0.004, r=−0.66).

The α-diversity (Chao1 richness and Shannon diversity index (H’) shown here) across a range of watersheds with different percentages of their area that had been mined (Observed, P=0.44; Chao1, P=0.74; and Shannon, P=0.37). The post hoc designations of sites (group A, group B and reference) are indicated in key.
We examined diversity variation among site categories using common biotic indices (Table 3). The α-diversity of bacteria OTUs did not differ between a priori designated site types using any of these indices (Kruskal–Wallis, P>0.05). However, post hoc categories did differ in α-diversity for Chao1 richness, Margalef’s index and evenness (Kruskal–Wallis, P=0.04, P=0.04, P=0.002), which was lower in group A (heavily mined) than group B (moderately mined) (Table 3).
Indicator taxa and predicted functions
We performed indicator species analysis of OTUs, orders and families to determine which taxa reliably indicated particular environmental conditions (Table 4). Comparing heavily mined, moderately mined and unmined sites, we found 174 OTU-based taxa strongly associated with one of these three groups. Most OTU indicators (n=156) closely associated with unmined sites, only 1 strongly associated with the moderately affected group B sites and 17 associated with the heavily affected group A. Of the OTUs assigned to a taxa identifier, we found 20 orders (of 128 total), 34 families (of 203 total) and 28 genera (of 255 total) that were indicator taxa for one of the three post hoc groups.
Out of all described taxa, percent of watershed mined explained significant linear trends in abundance for 9 of 72 classes, 18 of 128 orders and 12 of 203 families (Supplementary Table S4). Whereas the Acidobacteria-5 and Betaprotebacteria classes, YCC11 and Ellin329 orders, and Bacteriovoracaceae and EB1003 families correlated negatively with percent of watershed mined (all r>−0.6), the Acidimicrobiia class, Acidimicrobiales, SBR1032, and Rhodobacterales orders, and Phyllobacteriaceae, Methylophilaceae and Desulfobacteraceae families increased in relative abundance in streams of more heavily mined watersheds (all r>0.5).
Because cross-gradient patterns of taxa are often conducted at coarse levels of taxonomic resolution, we explored genera responses within the two most abundant classes: Betaproteobacteria, which negatively correlated with percent of watershed mined, and Alphaproteobacteria, which did not correlate with percent of watershed mined. For each genus within the class, we assessed abundance patterns across the gradient of percent mining using Quasi-Poisson regression (McCullagh and Nelder, 1989). The negative correlation between Betaproteobacteria relative abundance and percent of watershed mined did not hold for all genera within the class (Figure 6). Whereas 9 genera did show a negative response, 6 responded positively and 18 had no response. In Alphaproteobacteria, which showed no response to mining at the class level, 13 genera responded positively, 11 responded negatively and 28 showed no significant response (Figure 7). We referenced each responding genus with KEGG Organism modules and Bergey’s Manual (Garrity, 2005) to identify energy metabolisms that might respond to AlkMD constituents (Table 5). The majority of sulfur and nitrogen metabolism pathways were shared by both positive and negative responders. However, the only responsive genus reported to include a denitrifier increased with mining, whereas assimilatory sulfate reduction pathways were identified for genera who only responded negatively to mining.

Response to percent area of watershed mined by (a) class Betaproteobacteria; and genera within Betaproteobacteria: (b) Hydrogenophaga, a representative positive responder; (c) Rubrivivax, a representative non-responder; and (d) Polaromonas, a representative negative responder, using Quasi-Poisson regression.

Response to percent area of watershed mined by (a) class Alphaproteobacteria; and genera within Alphaproteobacteria: (b) Rhodobacter, a representative positive responder; (c) Sphingobium, a representative non-responde; and (d) Bradyrhizobium, a representative negative responder, using Quasi-Poisson regression.
Relative gene family abundances generated by the predicted functional profile using PICRUSt grouped into three level-2 functional categories that correlated negatively with percent watershed mined. Gene family relative abundances ranged from 11% to <1%. Percent watershed mined correlated negatively with gene families in ‘Signaling Molecules and Interaction’ (Environmental Information Processing category) (P=0.005, r=−0.59, 4% abundance), ‘Xenobiotics Biodegradation and Metabolism’ (Metabolism category) (P=0.01, r=−0.53, 0.4% abundance) and ‘Transport and Catabolism’ (Cellular Processes category) (P=0.02, r=−0.50, 0.2% abundance).
Discussion
Despite receiving extensive AlkMD contamination from the largest surface coal mine in Appalachia, microbial communities exposed to exceptionally high levels of AlkMD constituents in Mud River are no less diverse than nearby reference communities. Diversity in these streams best correlates with a multivariate factor that incorporates elevated biofilm Cd, Mn, Zn and Ni. Contrary to our predictions, overall bacterial diversity was not strongly correlated with the extent of upstream mining, although within mined sites, mining intensity correlated negatively with taxonomic richness. Despite only modest changes in α-diversity, we detected significant compositional differences between microbial communities of unmined and mining-affected streams. These compositional shifts resulted from changes in relative abundance rather than turnover between species at mined and unmined sites, as only 9% of OTUs differed enough between mined and unmined sites to serve as indicator taxa. There was limited evidence that these compositional shifts were driven by responses to nitrate and sulfate availability for use in energy metabolism. Rather, taxa shifts may be because of stressors that effect cellular processes and signaling.
Although our data suggest that bacterial community composition shifted with AlkMD exposure, we do not observe linear trends in α-diversity along the mining gradient because α-diversity varies widely at unexposed sites. However, within mined sites, richness decreased as more watershed area was mined. When an ecosystem undergoes extreme environmental alteration, such as mountaintop mining, we expect organisms favored by the changes to flourish and sensitive taxa to fail. This subsidy–stress response (Odum et al., 1979) can shift community composition across environmental gradients as well as increase diversity at intermediate exposure levels where sensitive and tolerant (or subsidized) taxa overlap (Niyogi et al., 2007). This is a possible explanation for microbial taxa richness and diversity responses to the AlkMD gradient. It is also consistent with a transplant experiment in the Clark Fork River drainage containing Butte copper mine where Feris et al. (2009) found bacterial taxa richness was greatest at low and moderate levels of metal contamination (As, Cd, Cu, Pb and Zn) and lowest in uncontaminated and highly contaminated sediments.
Many studies reporting microbial community responses to environmental contaminants occur in acid mine drainage systems (Baker and Banfield, 2003). Communities in these very acidic, metallic waters typically are less diverse than bacteria from neutral or alkaline streams (Lear et al., 2009; Kuang et al., 2013) and pH strongly correlates with phylogenetic diversity, richness and UniFrac distance (Kuang et al., 2013). Water column pH during our study period was not statistically distinct between mined and unmined sites and had no correlation with percent watershed mined, although it spanned nearly two orders of magnitude (6.9–8.6). AlkMD characteristically elevates pH (Griffith et al., 2012), and previous work at this field site found a strong positive correlation between percent watershed mined and pH (Lindberg et al., 2011). Unlike that earlier study of Mud River, this study occurred during winter/early spring at which time high flows dilute pH effects of mine drainage. In contrast to prior studies (Fierer and Jackson, 2006; Lauber et al., 2009; Rousk et al., 2010; Griffiths et al., 2011), pH was not an important correlate of composition or diversity metrics during this sampling period. This may be explained by the pHs we examined or the limited pH range in our study (2 orders of magnitude) relative to prior studies (⩾4 orders of magnitude). Yet, our results also contrast a study of diversity in streams of Hubbard Brook Experimental Forest in New Hampshire, USA, in which a similar range in pH (4–6.3) was shown to be the best correlate of microbial taxa richness across streams (Fierer et al., 2007). The lack of a pH diversity correlation in Mud River suggests that other chemical constituents were stronger determinants of bacterial community structure than pH, and this is perhaps unsurprising given the large difference in alkalinity, conductivity and numerous trace elements associated with surface mining.
Community composition differed significantly between unmined sites and sites downstream of surface mines. In mining-affected sites, we detected two distinct post hoc groups associated with different levels of AlkMD exposure. Bacterial communities in moderate AlkMD exposure sites were more diverse than communities with high contaminant exposure. The shift in composition between unmined and mined sites was best explained by elevated AlkMD constituents Ca2+, Li, SO42−, Se and Mg2+ and overall ionic strength. Composition differences between high and moderate AlkMD groups was best explained by greater biofilm Cd, Mn and Zn concentrations in the high-mining-affected sites. Earlier work shows that salinity and trace metals generate significant changes in microbial community composition (Baker and Banfield, 2003; Feris et al., 2003; Lozupone and Knight 2007; Giller et al., 2009; Auguet et al., 2010; Lami et al., 2013). A number of studies have found that Cd and Zn in particular alter microbial community composition (Ganguly and Jana, 2002; Sverdrup et al., 2006; Bouskill et al., 2010; Xie et al., 2011).
As previous studies recognize (Lozupone et al., 2007; Kuczynski et al., 2010), analysis methods influence conclusions about composition differences. Although both distance matrices that we used for creating NMDS ordinations yielded identical differences between site types, they also revealed unique patterns of community composition within site- types. GUniFrac incorporates phylogenetic relatedness into the distance matrix by more heavily weighting closely related taxa (Kuczynski et al., 2010; Chen et al., 2012). Because site-type differences in composition were less strong when using a GUniFrac rather than Bray–Curtis dissimilarity distance matrix, shifts in composition may be due to closely related taxa responding quite differently to AlkMD. This point is bolstered by the genera-level analyses that revealed that genera within the same class had varied responses to the mining gradient.
Ultimately, as our knowledge of microbial life history and physiology grows, we hope to map individual microbial traits onto phylogenies. Such knowledge would improve chemical pollution monitoring and predictions of microbial responses to ecosystem degradation. At present, mapping microbial traits to identities is highly limited, obscuring causes or consequences of specific compositional shifts in bacteria communities. Yet, examining taxa that respond strongly to AlkMD and comparing associated compositional changes with those across other environmental gradients may illuminate important microbial indicator taxa.
Taxonomic data sets are informative, but poorly resolved at genus and species levels (only 1.1% of OTUs were assigned to known species). Thus, it is a challenge to select appropriate taxonomic levels for best understanding microbial responses. At the class level, the strongest AlkMD responders were within Proteobacteria, Acidobacteria and Actinobacteria phyla. Similar to Feris et al. (2003), we found that Betaproteobacteria relative abundance decreased across a contamination gradient. Betaproteobacteria indicator taxa also correlate negatively with river and estuary water organic carbon content (Fortunato et al., 2013), and this may play a role in structuring AlkMD communities as dissolved organic carbon had a strong positive correlation with mining. In contrast, Feris et al. (2009) also found that in hyporheic sediments sourced from alkaline streams (pH 7.9–8.3), Alpha- and Gammaproteobacteria relative abundance increased with a metal contamination index. Although we used surface biofilms, not hyporheic sediments, we observed no such relationship. In our study, the strongest positive correlation with mining occurred in Actinobacteria that responded linearly rather than in a threshold manner.
After identifying taxa responsive to this gradient, the next step is investigating mechanisms and corresponding ecological implications. We anticipated that two ions that substantially increased with mining, sulfate and nitrate, would link to changes in taxa composition as these can be used in energy metabolism. Indeed, several AlkMD-tolerant taxa included Gamma- and Deltaproteobacteria, Nitrospira, Bacilli and Sphingobacteria, several of which perform biogeochemical transformations involving nitrogen cycling. These include nitrite oxidation to nitrate by Nitrospirae (Wakelin et al., 2008), methanol oxidation linked with denitrification by Methylophilaceae (Kalyuhznaya et al., 2009) and nitrate reduction and aromatic compound degradation by Rhodocyclales (Hesselsoe et al., 2009). However, KEGG Organism modules revealed similar nitrogen pathways for species in genera that increased and decreased with mining. Moreover, predicted gene family abundances for nitrogen metabolism were not positively correlated with mining. Sulfur metabolism had a similar outcome: predicted sulfur metabolism gene family abundances were not correlated with mining (and sulfate), yet the sulfate-reducing Desulfobacteraceae family increased in relative abundance in streams of more heavily mined watersheds. Because of the regime shift in many AlkMD-associated chemicals, it is likely that no single mechanism is responsible for the taxa patterns we observe. Rather, the multivariate nature of AlkMD is best represented by the percentage of watershed mined. This chemical regime shift may affect cellular processes and signaling as the predicted functional profile suggests. At the ecosystem level, it is possible that these effects could alter energy requirements, thus influencing carbon use efficiency and carbon cycling if energy is shunted toward cellular processes and away from growth. Nonetheless, it seems that the majority of functional categories are predicted to be redundant between bacterial communities spanning the mining gradient.
In conclusion, our study shows that stream biofilm bacterial composition in the Mud River system significantly differed between sites receiving AlkMD and unexposed sites. Average taxonomic richness in sites receiving moderate levels of AlkMD constituents exceeded that for unexposed or heavily exposed sites, creating a nonlinear relationship between exposure and diversity. At most taxonomic levels, few taxa were statistically dissimilar enough between exposure categories to indicate habitat specialization. The small number of strongly responding taxa and disparity in compositional similarity between GUnifrac and Bray–Curtis ordinations suggest that community shifts occur through families, genera and species rather than further up the hierarchy. Such results contrast macrofaunal responses to AlkMD exposure where entire orders of aquatic insects are lost from heavily AlkMD-affected streams (Pond et al., 2008; Pond, 2010, 2012). Testing microbial community functional responses is the next logical step toward understanding ecologically relevant links between compositional shifts and the strong chemical gradient AlkMD establishes in Central Appalachian streams.
References
Anderson MJ . (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol 26: 32–46.
Anderson MJ, Willis TJ . (2003). Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology 84: 511–525.
Auguet J-C, Barberan A, Casamayor EO . (2010). Global ecological patterns in uncultured Archaea. ISME J 4: 182–190.
Baker BJ, Banfield JF . (2003). Microbial communities in acid mine drainage. FEMS Microbiol Ecol 44: 139–152.
Barbour MT, Gerritsen J, Snyder BD, Stribling JB . (1999). Rapid Bioassessment Protocols for Use in Streams and Wadeable Rivers: Periphyton, Benthic Macroinvertebrates, and Fish, Second Edition. EPA 841-B-99-002. U.S. Environmental Protection Agency, Office of Water: Washington, D.C.
Bates ST, Berg-Lyons D, Caporaso JG, Walters WA, Knight R, Fierer N . (2011). Examining the global distribution of dominant archaeal populations in soil. ISME J 5: 908–917.
Beevers KR . (2006) Sampling Strategies for Particle Filtering SLAM. Troy, NY.
Bernhardt ES, Lutz BD, King RS, Fay JP, Carter CE, Helton AM et al. (2012). How many mountains can we mine? Assessing the regional degradation of Central Appalachian rivers by surface coal mining. Environ Sci Technol 46: 8115–8122.
Bernhardt ES, Palmer MA . (2011). The environmental costs of mountaintop mining valley fill operations for aquatic ecosystems of the Central Appalachians. Ann NY Acad Sci 1223: 39–57.
Bouskill NJ, Barker-Finkel J, Galloway TS, Handy RD, Ford TE . (2010). Temporal bacterial diversity associated with metal-contaminated river sediments. Ecotoxicology 19: 317–328.
Bray JR, Curtis JT . (1957). An ordination of the upland forest communities of Southern Wisconsin. Ecol Monogr 27: 325–349.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high- throughput community sequencing data. Nat Methods 7: 335–336.
Chao A . (1984). Nonparametric estimation of the number of classes in a population. Scand J Stat 11: 265–270.
Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, Wu GD et al. (2012). Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 28: 2106–2113.
De Beer D, Stoodley P . (2006). Microbial biofilms. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) SIAM Review vol. 52. Springer: New York, pp 904–937.
De Cáceres M, Legendre P . (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90: 3566–3574.
De Cáceres M, Legendre P, Moretti M . (2010). Improving indicator species analysis by combining groups of sites. Oikos 119: 1674–1684.
Dufrene M, Legendre P . (1997). Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol Monogr 67: 345–366.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Feris K, Ramsey P, Frazar C, Moore JN, Gannon JE, Holben WE . (2003). Differences in hyporheic-zone microbial community structure along a heavy-metal contamination gradient. Appl Environ Microbiol 69: 5563–5573.
Feris KP, Ramsey PW, Gibbons SM, Frazar C, Rillig MC, Moore JN et al. (2009). Hyporheic microbial community development is a sensitive indicator of metal contamination. Environ Sci Technol 43: 6158–6163.
Fierer N, Jackson RB . (2006). The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA 103: 626–631.
Fierer N, Morse JL, Berthrong ST, Bernhardt ES, Jackson RB . (2007). Environmental controls on the landscape-scale biogeography of stream bacterial communities. Ecology 88: 2162–2173.
Fortunato CS, Eiler A, Herfort L, Needoba JA, Peterson TD, Crump BC . (2013). Determining indicator taxa across spatial and seasonal gradients in the Columbia River coastal margin. ISME J 7: 1899–1911.
Ganguly S, Jana BB . (2002). Cadmium induced adaptive responses of certain biogeochemical cycling bacteria in an aquatic system. Water Res 36: 1667–1676.
Garrity G . (2005). The Proteobacteria. In Garrity G, Brenner DJ, Krieg NR, Staley JR (eds) Bergey’s Manual of Systematic Bacteriology. Springer: New York, pp 1106.
Giller KE, Witter E, McGrath SP . (2009). Heavy metals and soil microbes. Soil Biol Biochem 41: 2031–2037.
Griffith MB, Norton SB, Alexander LC, Pollard AI, LeDuc SD . (2012). The effects of mountaintop mines and valley fills on the physicochemical quality of stream ecosystems in the central Appalachians: a review. Sci Total Environ 417-418: 1–12.
Griffiths RI, Thomson BC, James P, Bell T, Bailey M, Whiteley AS . (2011). The bacterial biogeography of British soils. Environ Microbiol 13: 1642–1654.
Hering D, Buffagni A, Moog O, Sandin L, Sommerhauser M, Stubauer I et al. (2003). The development of a system to assess the ecological quality of streams based on Macroinvertebrates – design of the sampling programme within the AQEM project. Int Rev Hydrobiol 88: 345–361.
Hesselsoe M, Füreder S, Schloter M, Bodrossy L, Iversen N, Roslev P et al. (2009). Isotope array analysis of Rhodocyclales uncovers functional redundancy and versatility in an activated sludge. ISME J 3: 1349–1364.
Kalyuhznaya MG, Martens-Habbena W, Wang T, Hackett M, Stolyar SM, Stahl DA et al. (2009). Methylophilaceae link methanol oxidation to denitrification in freshwater lake sediment as suggested by stable isotope probing and pure culture analysis. Environ Microbiol Rep 1: 385–392.
Kirby CS, Cravotta CA . (2005). Net alkalinity and net acidity 2: practical considerations. Appl Geochem 20: 1941–1964.
Kuang J-L, Huang L-N, Chen L-X, Hua Z-S, Li S-J, Hu M et al. (2013). Contemporary environmental variation determines microbial diversity patterns in acid mine drainage. ISME J 7: 1038–1050.
Kuczynski J, Liu Z, Lozupone C, Mcdonald D, Fierer N, Knight R . (2010). Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat Methods 7: 813–819.
Lami R, Jones LC, Cottrell MT, Lafferty BJ, Ginder-Vogel M, Sparks DL et al. (2013). Arsenite modifies structure of soil microbial communities and arsenite oxidization potential. FEMS Microbiol Ecol 84: 270–279.
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31: 814–821.
Lauber CL, Hamady M, Knight R, Fierer N . (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75: 5111–5120.
Lear G, Niyogi D, Harding J, Dong Y, Lewis G . (2009). Biofilm bacterial community structure in streams affected by acid mine drainage. Appl Environ Microbiol 75: 3455–3460.
Lindberg TT, Bernhardt ES, Bier R, Helton AM, Merola RB, Vengosh A et al. (2011). Cumulative impacts of mountaintop mining on an Appalachian watershed. Proc Natl Acad Sci USA 108: 20929–20934.
Logares R, Lindström ES, Langenheder S, Logue JB, Paterson H, Laybourn-Parry J et al. (2012). Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J 7: 1–12.
Lozupone CA, Hamady M, Kelley ST, Knight R . (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73: 1576–1585.
Lozupone CA, Knight R . (2007). Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104: 11436–11440.
McCullagh P, Nelder J . (1989) Generalized Linear Models 2nd edn Chapman and Hall/CRC: Boco Raton, pp 511.
McCune B, Mefford MJ . (2011). PC-ORD. Multivariate Analysis of Ecological Data. Gleneden Beach, Oregon, USA: MjM Software.
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M et al. (2008). The metagenomics RAST server — a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9: 386.
Niyogi DK, Koren M, Arbuckle CJ, Townsend CR . (2007). Stream communities along a catchment land-use gradient: subsidy-stress responses to pastoral development. Environ Manage 39: 213–225.
Odum EP, Finn JT, Franz EH . (1979). Perturbation theory and the subsidy-stress gradient. Bioscience 29: 349–352.
Paerl HW, Dyble J, Moisander PH, Noble RT, Piehler MF, Pinckney JL et al. (2003). Microbial indicators of aquatic ecosystem change: current applications to eutrophication studies. FEMS Microbiol Ecol 46: 233–246.
Palmer MA, Bernhardt ES, Schlesinger WH, Eshleman KN, Foufoula-Georgiou E, Hendryx MS et al. (2010). Mountaintop mining consequences. Science 327: 148–149.
Pond GJ . (2010). Patterns of Ephemeroptera taxa loss in Appalachian headwater streams (Kentucky, USA). Hydrobiologia 641: 185–201.
Pond GJ . (2012). Biodiversity loss in Appalachian headwater streams (Kentucky, USA): Plecoptera and Trichoptera communities. Hydrobiologia 679: 97–117.
Pond GJ, Passmore ME, Borsuk FA, Reynolds L, Rose CJ . (2008). Downstream effects of mountaintop coal mining: comparing biological conditions using family- and genus-level macroinvertebrate bioassessment tools. J North Am Benthol Soc 27: 717–737.
Rose AW, Cravotta CAI . (1998). Geochemistry of coal mine drainage. In: Brady KBC, Smith MW, Schueck J (eds) Coal Mine Drainage Prediction and Pollution Prevention in Pennsylvania. Pennsylvania Department of Environmental Protection: Harrisburg, PA, pp 1–22.
Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG et al. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4: 1340–1351.
Sabater S, Guasch H, Ricart M, Romaní A, Vidal G, Klünder C et al. (2007). Monitoring the effect of chemicals on biological communities. The biofilm as an interface. Anal Bioanal Chem 387: 1425–1434.
Sims A, Zhang Y, Gajaraj S, Brown PB, Hu Z . (2013). Toward the development of microbial indicators for wetland assessment. Water Res 47: 1711–1725.
Solimini AG, Free G, Donohue I, Irvine K, Pusch M, Rossaro B et al. (2006). Using benthic macroinvertebrates to assess ecological status of lakes current knowledge and way forward to support WFD implementation. Institute for Environment and Sustainability, European Commission.
Sverdrup LE, Linjordet R, Strømman G, Hagen SB, van Gestel CAM, Frostegård S et al. (2006). Functional and community-level soil microbial responses to zinc addition may depend on test system biocomplexity. Chemosphere 65: 1747–1754.
Townsend P, Helmers D, Kingdon C, Mcneil B, Debeurs K, Eshleman K . (2009). Changes in the extent of surface mining and reclamation in the Central Appalachians detected using a 1976–2006 Landsat time series. Remote Sens Environ 113: 62–72.
USEPA. (2011) The Effects of Mountaintop Mines and Valley Fills on Aquatic Ecosystems of the Central Appalachian Coalfields. U.S. Environmental Protection Agency: Washington, DC.
USGS (Variously dated). National field manual for the collection of water-quality data U.S. Geological Survey Techniques of Water-Resources Investigations book 9, chaps. A1-A9, available online at http://pubs.water.usgs.gov/twri9A.
Wakelin SA, Colloff MJ, Kookana RS . (2008). Effect of wastewater treatment plant effluent on microbial function and community structure in the sediment of a freshwater stream with variable seasonal flow. Appl Environ Microbiol 74: 2659–2668.
Wetzel RG . (1983) Limnology 2nd edn Saunders College Publishing: Philadelphia, PA, USA.
Xie X, Liao M, Ma A, Zhang H . (2011). Effects of contamination of single and combined cadmium and mercury on the soil microbial community structural diversity and functional diversity. Chin J Geochem 30: 366–374.
Acknowledgements
We thank Ashley Helton, Ty Lindberg, Brooke Hassett, Anna Fedders, Mariah Arnold and Yu-Ting Liu for research assistance and two anonymous reviewers for comments. We thank the Miller family for sampling location guidance. Funding was provided by Foundation for the Carolinas to Nicholas School of the Environment, Duke University, as an unrestricted gift.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Bier, R., Voss, K. & Bernhardt, E. Bacterial community responses to a gradient of alkaline mountaintop mine drainage in Central Appalachian streams. ISME J 9, 1378–1390 (2015). https://doi.org/10.1038/ismej.2014.222
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.222
This article is cited by
-
Mine reclamation does not restore leaf processing in low-order streams
Hydrobiologia (2023)
-
Pollution shapes the microbial communities in river water and sediments from the Olifants River catchment, South Africa
Archives of Microbiology (2021)
-
Exploring the allochthonous pollution influence on bacterial community and co-occurrence dynamics of River Ganga water through 16S rRNA-tagged amplicon metagenome
Environmental Science and Pollution Research (2021)
-
How sediment bacterial community shifts along the urban river located in mining city
Environmental Science and Pollution Research (2021)
-
Bacterial community composition and potential pathogens along the Pinheiros River in the southeast of Brazil
Scientific Reports (2020)
