
Abstract
Microbial interactions have a key role in global geochemical cycles. Although we possess significant knowledge about the general biochemical processes occurring in microbial communities, we are often unable to decipher key functions of individual microorganisms within the environment in part owing to the inability to cultivate or study them in isolation. Here, we circumvent this shortcoming through the use of single-cell genome sequencing and a novel low-input metatranscriptomics protocol to reveal the intricate metabolic capabilities and microbial interactions of an alkane-degrading methanogenic community. This methanogenic consortium oxidizes saturated hydrocarbons under anoxic conditions through a thus-far-uncharacterized biochemical process. The genome sequence of a dominant bacterial member of this community, belonging to the genus Smithella, was sequenced and served as the basis for subsequent analysis through metabolic reconstruction. Metatranscriptomic data generated from less than 500 pg of mRNA highlighted metabolically active genes during anaerobic alkane oxidation in comparison with growth on fatty acids. These data sets suggest that Smithella is not activating hexadecane by fumarate addition. Differential expression assisted in the identification of hypothetical proteins with no known homology that may be involved in hexadecane activation. Additionally, the combination of 16S rDNA sequence and metatranscriptomic data enabled the study of other prevalent organisms within the consortium and their interactions with Smithella, thus yielding a comprehensive characterization of individual constituents at the genome scale during methanogenic alkane oxidation.
Similar content being viewed by others

Introduction
Smithella, a member of the family Syntrophaceae, has been shown to be a predominant member of microbial communities involved in the anoxic degradation of long-chain alkanes (Zengler et al., 1999; Gray et al., 2011; Siddique et al., 2011, Wang et al., 2011; Scherr et al., 2012; Cheng et al., 2013). The mechanism driving anoxic hexadecane degradation in methanogenic consortia is thus far unknown. However, it is likely that this conversion requires the interaction of syntrophic bacteria, such as members of the Syntrophaceae family, with methanogenic archaea (Zengler et al., 1999). The scope of previous studies has been limited to identifying Syntrophaceae as key organisms within complex communities facilitating the initial steps of alkane activation (Gieg et al., 2008; Gray et al., 2011; Aitken et al., 2013). This can be attributed to both the difficulty in isolating pure cultures of members of the Smithella genus as well as the slow growth and low biomass of the culture, with most experiments taking hundreds of days to complete (Figure 1). To date, only one member of the genus Smithella, Smithella propionica, has been isolated as an axenic culture (Liu et al., 1999).

(a) Formation of gas (methane) in an anaerobic enrichment culture growing in 300 ml mineral medium containing hexadecane (•). The methanogenic consortium was propagated in the laboratory and formed a total of 809 ml of gas consisting of 10.9 mM methane (8.81 mmol total formed) after 1141 days of incubation. A control without hexadecane (▪) showed no gas formation. The previous enrichment described in Zengler et al., 1999 (open symbols, overlaid for comparison) formed a total of 370 ml of gas after 1051 days of incubation. The community with hexadecane is represented by open circles (∘), and the control by open squares (□). (b) The consortium, although highly metabolically active as seen by gas formation (bubbles), grows to very low biomass densities as can be seen in this 3-year-old culture. Teflon boiling stones were coated with hexadecane to increase the contact between cells and the hydrophobic substrate.
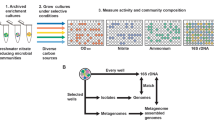
Elucidating the genetic basis of mechanisms driving the observed phenotype of unculturable microbial communities is contingent upon the deconvolution of the roles of individual community constituents (Zengler and Palsson, 2012). Owing to recent advances in next-generation sequencing, the study of non-model organisms in microbial communities is becoming increasingly accessible. Whole-genome sequencing of individual species within communities is routinely performed without the need for cultivation (Swan et al., 2011; Yoon et al., 2011; Seth-Smith et al., 2013). Furthermore, unlike hybridization-based approaches, entire transcriptomes can be characterized without the knowledge of existing reference genomes before sequencing (Wang et al., 2009). Here, we study individual unculturable constituents in a low-biomass microbial community degrading hexadecane to methane (Figure 2). We applied a single-cell genome sequencing approach to assemble a working-draft genome of Smithella, and subsequently used it to identify potential alkane degradation-related genes and to reconstruct metabolic pathways. We deployed a low-input metatranscriptomic approach to accommodate the low biomass of the culture in order to determine which genes were active during alkane oxidation. After determining the major role of Smithella within the community, the metatranscriptomic data sets were extended to characterize the activity of other microbial community members cultured with a variety of substrates, in an effort to elucidate the microbial interactions that mediate community composition fluctuations during growth with different carbon sources.

Schematic of the integrated workflow applied to study the methanogenic community. A single-cell genome sequencing approach established a working-draft genome of Smithella. Then, low-input metatranscriptomics was used in order to determine which genes were active during alkane degradation. After determining the major role of Smithella within the community, the metatranscriptomics data sets were extended to analyze the activity of other microbial community members. A genome-scale metabolic model was used to facilitate the integration of both the genomic and transcriptomic data in order to extract functional information about the organisms.
Materials and methods
Media and cultivation
The original source of the enrichment was sediment from a hydrocarbon-contaminated ditch in Bremen, Germany (Zengler et al., 1999). The consortium was further propagated in the laboratory in an anoxic medium containing 0.3 g NH4Cl, 0.5 g MgSO4·7H2O, 2.5 g NaHCO3, 0.5 g K2HPO4, 0.05 g KBr, 0.02 g H3BO3, 0.02 g KI, 0.003 g Na2WO2·2H2O, 0.002 g NiCl2·6H2O, trace elements and trace minerals as previously described (Zengler et al., 1999). The medium was sparged with a mixture of N2/CO2 (80:20 v/v), and the pH was adjusted to 7.0. After autoclaving, anoxic CaCl2 (final concentration 0.25 g l−1) and filter-sterilized vitamin solution (Zengler et al., 1999) were added. Cells were supplemented with anoxic hexadecane (0.5 ml, equaling 1.7 mmol, on a teflon filter, see Zengler et al, 1999), 5 mM butyric acid or 2 mM caprylic acid and incubated at 30 °C in triplicate. Bottles were degassed as necessary to relieve overpressurization. The headspace of a representative active culture was analyzed using gas chromatography (Shimadzu GC 2014, Supelco 30 m × 0.53 mm Carboxen column, Kyoto, Japan). A thermal conductivity detector was used to identify and quantify the production of methane accompanying cell growth. Ultra pure nitrogen was used as the carrier gas, and the column was run at 35 °C for 15 min with a flow rate of 20 ml min−1.
Single-cell sorting, multiple displacement amplification and genome sequencing
Individual cells from a single bottle of the methanogenic alkane-degrading consortium (Figure 1) were obtained by staining the cells with SYTO-9 DNA stain and sorting of single cells using fluorescence-activated cell sorting. Single cells were then lysed as previously described, and the genomic DNA of individual cells was amplified using whole-genome multiple displacement amplification (Swan et al., 2011). Amplified genomic DNA was screened for Smithella-specific 16S rDNA gene sequences. Six amplified Smithella genomes were selected for next-generation sequencing. The multiple displacement amplified genomes were prepared for Illumina sequencing using the Nextera kit, version 1 (Illumina, San Diego, CA, USA) using the Nextera protocol (ver. June 2010) and a high-molecular-weight buffer. Libraries of these six samples were created and sequenced using an Illumina Genome Analyzer II, generating a total of 174 million paired-end reads using 36 or 58 bp reads with an insert size of 400 (Supplementary Table S1).
Assembly of single-cell genomes
Sequenced reads from Smithella were assembled using the de novo co-assembler, HyDA (Movahedi et al., 2012, http://compbio.cs.wayne.edu/software/hyda/). HyDA is an assembler based on the colored de Bruijn graph. In the beginning, a unique color is assigned to each input data set, which belongs to one species. Here, the Smithella genome was co-assembled with the single-cell genomic material from two additional species (Anaerolina sp and Syntrophus sp) from the culture using three colors. All the colored input data sets were assembled simultaneously in a unified de Bruijn graph. The output of HyDA is a list of contigs and the corresponding average sequencing depth (coverage) for each color/input data set. Zero average coverage indicates the absence of that contig in the assembled genome of the corresponding data set. Unicolored contigs are pieces of a sequence that are unique to the corresponding data set, and those contigs that have non-zero coverage for multiple data sets are shared sequences. The output of HyDA is the collective list of contigs present in at least one of the input data sets. To run the software, we chose the length of k-mers in the de Bruijn graph to be 25, and the coverage cutoff to trim erroneous branches in the graph was selected to be 100. The contigs were then annotated using RAST (rapid annotations using subsystem technology) (Aziz et al., 2008), and the resulting annotation was used to generate a draft metabolic reconstruction using ModelSEED (Henry et al., 2010). This Whole-Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession number AWGX00000000. The version described in this paper is version AWGX01000000.
Low-input metatranscriptomics
To identify metabolically active genes, the transcriptome of the community was sequenced. Twenty milliliters of culture was harvested (from the same bottle used for single-cell genome sequencing) by adding 2 ml stop solution (95% ethanol, 5% TRIzol (Life Technologies, Carlsbad, CA, USA)) to the sample and mixing by inversion. For the fatty acid cultures, cells were harvested after 171 days of incubation (Supplementary Figure S1). Hexadecane samples were harvested 1146 days (3.14 years) after inoculation. The activity of the culture was monitored by measuring the amount of gas and methane produced, as OD600 proved to be an inaccurate measure owing to the aggregation of cells within the community. The hexadecane-degrading consortium had produced 180 ml of gas at the time of harvest. Cells were then centrifuged at 10 000 g at 4 °C for 10 min in 50 ml conical tubes; centrifugation at lower r.p.m. (8000 r.p.m.) resulted in an insufficient recovery of Smithella sequences. After the initial pelleting, the supernatant was removed and centrifuged again in 1.5 ml-microcentrifuge tubes at 14 000 g at 4 °C for an additional 20 min. After centrifugation, the supernatant was decanted and cell pellets were frozen at −80 °C until use. Both pellets were combined before RNA isolation.
RNA was isolated, and rRNA was removed using a modified version of the Metabacteria RiboZero rRNA Removal kit (Epicentre, Madison, WI, USA). cDNA was generated using purified mRNA, and sequencing libraries were constructed using a modified version of the Nextera protocol (Illumina). A more detailed version of the protocol is given in the Supplemental Methods.
To benchmark the low-input protocol against standard RNA-seq methods, the standard dUTP method (Parkhomchuk et al., 2009) as well as the low-input method were applied to a Geobacter sulfurreducens and G. metallireducens coculture (Shrestha et al., 2013).
Mapping of transcriptome reads
The metatranscriptomics libraries generated were sequenced using either a Genome Analyzer IIx or a MiSeq (Illumina). The obtained reads were mapped to the Smithella draft genome using the short-read aligner Bowtie (http://bowtie-bio.sourceforge.net) (Langmead et al., 2009) with two mismatches allowed per read alignment. To estimate transcript abundances, FPKM values were calculated using the Cufflinks tool (http://cufflinks.cbcb.umd.edu/) (Trapnell et al., 2010). Differential expression analysis was performed for the community under hexadecane-oxidizing conditions compared with butyric acid- and caprylic acid-oxidizing conditions using the Cuffdiff feature of the cufflinks package. DESeq (Anders and Huber, 2010) was also used to confirm the differential expression observed in the alkane-oxidizing community. The number of reads mapped to each gene was determined using HTSeq (Anders S, http://www-huber.embl.de/users/anders/HTSeq/). Differential expression analysis was also calculated for controls. In each case, the library prepared using the standard dUTP method was compared with the library generated by the low-input method described here.
In order to analyze the transcript abundance of genes not present in a specific genome, a de novo assembly of the community metatranscriptome under the hexadecane-degrading conditions was constructed using Trinity (Grabherr et al., 2011). The de novo metatranscriptome was annotated using RAST (Aziz et al., 2008) and served as a reference for mapping (Langmead et al., 2009). HTSeq (Anders S, http://www-huber.embl.de/users/anders/HTSeq/) was used to generate count data for specific genes. Metatranscriptome data analysis can be accessed through GEO, accession no. GSE49830.
16S rDNA sequencing and analysis
DNA was isolated from the community cultured under hexadecane-, butyric acid- and caprylic acid-degrading conditions using Nucleospin XS Tissue columns (Clontech, Mountain View, CA, USA) using the same lysis modifications described in the Supplemental ‘Low-Input Metatranscriptomics’ section. In place of Superase-In, 1 μl RNAse A (Qiagen, Venlo, Netherlands) was added to cultures, and cells were lysed for 1.5 h. Long primers containing Illumina-compatible adapters were used to amplify bacterial-specific (27F/806R) and archaeal-specific (21F/806R) V1 and V4 regions of the 16S rDNA gene. The PCR product was purified using 0.7 × AMPure beads, and confirmed using a Bioanalyzer DNA High Sensitivity chip (Agilent, Santa Clara, CA, USA). Regions V1-V2 (read 1) and V4 (read 2) were sequenced on a Miseq (Illumina) using 250 bp on each end. Reads one and two were processed as replicates using QIIME. Standard parameters were used for operational taxonomic unit selection, and taxonomy was assigned using the most recent version (ver. 12_10) of Greengenes. Operational taxonomic units with only one read were filtered from the results before analysis.
Results
Single-cell genome sequencing and assembly
The Smithella de novo assembly generated with HydA resulted in 7697 contigs totalling to 3 213 255 bp (Table 1). The N50 was 6198 bp, and the largest contig was 56 011 bp. These contigs were used as a draft genome for Smithella for all downstream analysis. To estimate the completeness of this draft genome, annotated proteins within the genome were compared with clusters of orthologous groups (COGs), categories identified for Syntrophus aciditrophicus (strain SB, accession no. CP000252). As to date no members of the genus Smithella have been sequenced, S. aciditrophicus, a close relative of Smithella (Supplementary Figure S2) whose genome has been sequenced, was chosen (McInerney et al., 2007). In all, 2250 of the 2863 (79%) annotated Smithella genes were functionally categorized into COGs, with most COGs being represented above 80% compared with S. aciditrophicus (Supplementary Table S2). The number of genes falling into the category ‘lipid transport and metabolism’ is twice as large (209%), as expected based on the S. aciditrophicus COGs, hinting at the capability of Smithella to metabolize fatty acids, which are potential intermediates of anaerobic alkane oxidation (Rojo, 2009).
After annotating the Smithella draft genome, ModelSEED was used to generate a draft metabolic reconstruction and to group genes into corresponding metabolic pathways (Henry et al., 2010). In total, 510 genes were grouped into 731 different metabolic reactions. These reactions encompassed 229 different subsystems (Supplementary Figure S3), including a number of potential pathways related to alkane and fatty acid metabolism. Next, the reconstruction of Smithella was compared with a ModelSEED reconstruction of S. aciditrophicus to benchmark the completeness of the functional Smithella genome and to identify potential pathways involved in alkane degradation. Only genes that were part of complete, functional subsystems were included in the comparison. Of the 1152 genes included in the comparison, 136 were found only in Smithella whereas 453 were S. aciditrophicus-specific (Supplementary Table S3). Many of the S. aciditrophicus-specific genes fell into the categories DNA metabolism (42) and motility and chemotaxis (36), consistent with our COG analysis that suggests that the categories ‘replication, recombination and repair’ and ‘cell motility’ are less prevalent in the Smithella draft genome. Seven fatty acid-related genes were identified as Smithella-specific, as well as four genes related to cytochrome biosynthesis.
Genes expressed during alkane degradation and comparative transcriptomics of Smithella
In order to determine what functional genes are active under hexadecane-degrading conditions, we evaluated the transcriptome of Smithella. Standard protocols for RNA isolation and sequencing proved to be ineffective owing to the very low biomass of the community. We therefore adapted a low-input metatranscriptomics protocol to accommodate the low biomass of the culture (Figure 1b). The validity and applicability of this protocol to other microbial communities were assessed by analyzing mock communities in biological duplicate using both the low-input method presented here as well as a standard dUTP method (Parkhomchuk et al., 2009). The mock community consisted of a bacterial coculture of G. sulfurreducens and G. metallireducens (Shrestha et al., 2013). Libraries created via the low-input method utilized 500 pg of starting mRNA, whereas those created according to the dUTP method used 100 ng of starting mRNA. Sequenced reads from the Geobacter coculture were mapped to both organisms, and differential expression between the low-input and dUTP libraries for each sample was calculated using the Cufflinks package (Trapnell et al., 2010). No significant, differential gene expression was observed between the two library preparation methods. Gene expression values were also correlated to assess overall transcript representation within each library type (Supplementary Figure S4). The Spearman rank correlation of replicates 1 and 2 was 0.9 and 0.86, respectively.
Using this low-input protocol, a metatranscriptomic library of the methanogenic community grown with hexadecane as the sole electron donor was generated using ∼500 pg of mRNA. The library was sequenced on an Illumina GAIIx, generating 53 million reads (5.3 Gb of data), of which 480 Mb mapped to the Smithella draft genome. Interspecies sequence diversity of community members combined with high stringency settings during mapping ensured that only reads derived from Smithella were included in downstream gene expression analysis. Our mapping strategy only permitted two mismatches as well as alignment suppression if one reportable alignment already existed for a particular read. This allowed us to isolate a Smithella-specific transcriptome profile out of the metatranscriptome (Figure 3). The transcriptomic reads covered 94% of all genes in the 3.2-Mb Smithella draft genome. A number of putative genes related to alkane degradation, including potential alkane-emulsification genes and fatty acid degradation genes, were identified as highly expressed (>75th percentile) (Supplementary Table S5) while Smithella metabolizes hexadecane.

Example of the coverage obtained from the metatranscriptome (hexadecane-degrading community) when mapped to the Smithella draft genome (teal tracks) and the published genome of M. concilii (orange tracks). This snapshot is representative of the level of coverage observed across the entire genome of these organisms. Ninety-four percent of the genes from Smithella were represented by the metatranscriptomic data set.
To discriminate between genes related to alkane and fatty acid metabolism, differential expression of the community utilizing hexadecane, butyric or caprylic acid was explored (Supplementary Table S4, Supplementary Figure S1). Metatranscriptomic reads from these two fatty acid communities were then mapped to the Smithella draft genome, and from this data set, we identified genes of Smithella that were actively expressed only in the presence of hexadecane. Potential alkane-activation genes were of particular interest. A number of radical-activating enzymes that exhibited homology to gene sequences in known, anaerobic hexadecane-degraders including Desulfatibacillum alkenivorans (Callaghan et al., 2012) and Desulfococcus oleovorans HDX3 (Aeckersberg et al., 1991), were identified as highly expressed (all but one within the upper 25th percentile) (Supplementary Table S7). Because Smithella may be degrading hexadecane through an unknown pathway, the homology of hypothetical proteins (68) falling into the top 10% of all expressed genes during hexadecane degradation were also investigated. Hypothetical proteins exhibiting high levels of expression across all three conditions were excluded from the analysis. Twenty one out of 68 candidate proteins were found to have high homology (position-specific iterative basic local alignment score ⩾200) to hypothetical proteins encoded in the genomes of hexadecane-degraders D. alkenivorans and D. oleovorans (Supplementary Table S8). The remaining 47 proteins had no homology to known proteins. Because all of these genes were highly expressed, future biochemical analysis of these gene products is necessary to elucidate if these genes are directly involved in hexadecane degradation.
Community composition and interactions during alkane and fatty acid degradation
Elucidating the metabolic activity of Smithella subsequently allowed determining potential interspecies interactions within the methanogenic community. Bacterial and archaeal 16S rDNA sequence analysis revealed major shifts in the community composition between growth with hexadecane, butyric acid or caprylic acid (Figure 4b). While Smithella comprised 82% of the community during decomposition of hexadecane, its abundance dropped to 17% when butyric or caprylic acid was the energy source. In their stead, members of the families Dethiosulfovibrionaceae (29% and 19%, butyric acid and caprylic acid, respectively) and Desulfobulbaceae (20%) began to dominate the community. Unknown members of the orders Ignavibacteriales (10%) and Bacteroidales (13% and 11%) also became more predominant during growth on fatty acids. Within the archaea portion of the community, Methanosaeta was the prevalent methanogen under two conditions. It comprised 93% of the archaeal community when butyric acid was provided, 83% for caprylic acid, but only 37% for hexadecane. Sequences related to Methanoculleus (Methanomicrobiales) were found in all conditions at levels below 10%. Under hexadecane-degrading conditions, Methanocalculus, another member of the Methanomicrobiales, represented 47% of all archaea (Figure 4b).

(a) Overview of the metabolism of Smithella and two representative methanogens during hexadecane degradation. Active pathways were determined from metatranscriptomic data mapped to the Smithella draft genome and the published M. concilii genome. The red arrow indicates that the mechanism driving this step is currently unknown. In this paper, we present a list (Supplementary Tables S7 and S8) of highly expressed, hypothetical proteins and radical-activating enzymes that may be facilitating hexadecane activation. (b) 16S rDNA gene analysis of the bacterial and archaeal community under hexadecane-, caprylic acid- and butyric acid-degrading conditions.
Metatranscriptomic data sets were mapped to the genomes of the closest sequenced members identified from the 16S rDNA data to determine metabolically active members of the community. We mapped reads to five Deltaproteobacteria and thirteen different representative methanogens within the orders Methanobacteriales, Methanosarcinales and Methanomicrobiales (Table 2, Supplementary Table S4).
Activity of Methanosaeta concilii
Within the hexadecane-degrading culture, 268 Mb mapped to a species related to M. concilii, consistent with 16S rDNA data suggesting Methanosaeta is an active member of the community. Genes related to energy metabolism and methanogenesis were highly expressed across all conditions. Of particular note was the expression of genes involved in CO2 fixation (Figure 4a, Supplementary Table S6). Nearly all subunits of the major genes involved in the CO2 fixation pathway were expressed during hexadecane degradation with transcript abundance above the median FPKM (168 for butyric acid comparison and 359 for caprylic acid comparison). Only formyl-MF subunits fmdABCF were expressed below the median FPKM, as well as formyl-MF:tetrahydromethanopterin formyltransferase. Similarly, most subunits of acetyl-CoA synthetase and CO dehydrogenase/acetyl-CoA synthase, the key enzymes facilitating methanogenesis from acetate, were expressed above the median FPKM with only cdhA and cdhE being expressed below the median FPKM. The acetate transporter, MCON_2287, was expressed below the median FPKM.
Activity of Methanosaeta harundinacea
While almost no reads (1.4 Mb) mapped to M. harundinacea (another Methanosaeta species) during degradation of hexadecane, a significant portion of reads mapped to this species during fatty acid degradation with 58 Mb mapping for butyric acid and 31 Mb mapping for caprylic acid (Supplementary Table S4). Although the reads mapped evenly to the M. concilii genome, the mapping to M. harundinacea was uneven despite the high overall number of reads aligning to the genome. This discrepancy is likely due to sequence dissimilarities between the published M. harundinacea genome and the species of Methanosaeta within the culture presented here. Thus, instead of analyzing relative expression levels of genes within a specific condition, count data were used to investigate potentially active genes across all three conditions, as the same regions of the genome should be covered during read mapping. The genes with the highest hits, particularly during caprylic acid degradation, were formate dehydrogenase genes (Mhar_0689: 1678, Mhar_1941: 1151, Mhar_0325: 1084, Mhar_1279: 706 counts) as well as the CO2 fixation-gene formyl-MF dehydrogenase (Mhar_1287: 697 counts) and the acetate-fermentation genes acetyl-CoA synthetase (Mhar_0749: 967 counts) and acetyl-CoA synthase/decarbonylase (Mhar_2328: 735 counts).
Activity of Methanoculleus marisnigri
Reads of cultures growing with butyric or caprylic acid also mapped to M. marisnigri, a hydrogenotrophic methanogen. In contrast to M. concilli, this organism is incapable of growing with acetate as the sole electron donor (Maestrojuán et al., 1990). A total of 197 Mb mapped to M. marisnigri under butyric acid (across 1022 of 2557 genes) and 113 Mb (across 872 of 2557 genes) under caprylic acid-degrading conditions (no reads from hexadecane growth mapped to M. marisnigri). Again, genes involved in methanogenesis, such as methyl-coenzyme M reductase and formyl-MF:tetrahydromethanopterin formyltransferase, were highly expressed. However, as a reference genome was used instead of a genome sequence actually obtained from a cell in the community, reads were not efficiently mapped to all genes, likely due to inherent sequence dissimilarities between species as was observed for M. harundinacea.
Activity of Methanocorpusculum labreanum (Methanocalculus)
No reads under any condition could be mapped to M. labreanum, the only member of the Methanocorpusculaceae that has been sequenced so far. This family represented 47% of all archaea within the hexadecane community, but only less than 10% in the two fatty acid-metabolizing communities, hinting at a specific role of these bacteria related to hexadecane degradation. To try and determine the role of this organism within the community, expression of formate dehydrogenases and the energy conserving hydrogenases (ech) was investigated through the use of a de novo assembled metatranscriptome. Under hexadecane-degrading conditions, 4069 reads mapped to formate dehydrogenase genes and 579 to ech hydrogenase genes, whereas only 42 and 47 reads mapped to formate dehydrogenase genes and 6 and 318 reads mapped to ech hydrogenase genes in the butyric acid and caprylic acid communities, respectively (Supplementary Table S9). This suggests that both formate dehydrogenases and ech hydrogenases are highly active during hexadecane degradation compared with growth on fatty acids. The lowered transcript counts may also reflect the low Methanocalculus abundance in the fatty acid conditions.
Discussion
Obtaining information about the activity of individual members within a microbial community with respect to environmental perturbation, such as availability of different electron donors or acceptors, has long been stymied by the inability to link metatranscriptomic data to specific microorganisms. Metatranscriptomic data were generated for the community growing on hexadecane, butyric acid or caprylic acid using a low-input metatranscriptomics protocol optimized for the very low biomass of the community (Figure 1b). These metatranscriptomes were used in conjunction with the Smithella draft genome, obtained from single-cell sequencing and other representative genomes, to investigate both metabolic capabilities and microbial interactions of the community.
In Smithella, a number of pathways potentially related to alkane degradation were identified in the genome. Comparison of transcriptomic data obtained from all three carbon source additions explicated the activity of these genes during hexadecane degradation. Emulsification and membrane genes that are known to assist in the degradation of hydrophobic alkanes (Tzintzun-Camacho et al., 2012) were both present and active within Smithella. The gene encoding an apolipoprotein N-acyltransferase is of particular note, as previous studies have shown that deletion of lipoprotein synthesis genes can decrease membrane hydrophobicity in bacteria (Okugawa et al., 2012). Expression of this gene during growth with hexadecane may assist Smithella to adapt its membrane composition to the presence of the hydrophobic substrate. Transporters for long-chain fatty acids, such as palmitate, octadecanoate and tetradecanoate, were also encoded in the genome and actively expressed during growth with hexadecane only, suggesting that these genes may be assisting in the export of biosurfactants for hexadecane emulsification (Breuil and Kushner, 1980; Liu et al., 2012).
To date, very few enzymes involved in the activation of alkanes without molecular oxygen have been elucidated (So et al., 2003; Zedelius et al., 2011; Callaghan et al., 2012). Although the Smithella genome enodes a gene for methylmalonyl-CoA mutase, a protein known to have a role in fumarate-dependent anaerobic hexadecane degradation (Wilkes et al., 2002; Callaghan et al., 2012), this mode of degradation seems unlikely in this organism. Genes for alkyl-succinate synthase subunits, critical for the fumarate-dependent activation of alkanes, were not identified in the genome. This is consistent with previous studies that suggest a mechanism other than fumarate addition that drives alkane activation under methanogenic conditions (Aitken et al., 2013). Genes highly expressed during growth with hexadecane and not with fatty acids, particularly hypothetical proteins with no annotated function (Supplementary Tables S7 and S8), could provide potential candidates involved in this pathway.
Biological conversion of hexadecane to methane requires the interaction of syntrophic bacteria with methanogenic archaea (Zengler et al., 1999). A number of methanogens, notably the acetoclastic M. concilii and M. harundinacea as well as the hydrogenotrophic M. marisnigri and Methanocalculus were found to be highly abundant in our community. On the basis of 16S rDNA data, Methanosaeta species were well represented under all three different growth conditions, but dominated during fatty acid degradation (Figure 4b). While M. concilii was almost exclusively active during hexadecane degradation based on transcriptomic analysis, both M. concilii and M. harundinacea were active during oxidation of fatty acids (Supplementary Table S4). Although at least two different species of Methanosaeta appeared to be active within the community, they are fulfilling different metabolic roles under each condition. M. harundinacea is likely using formate as an electron donor in addition to acetate utilization during fatty acid degradation. The M. concilii genome does not encode a complete formate–dehydrogenase complex. Thus, if formate is being used as an electron carrier during hexadecane degradation as was revealed through de novo transcriptome analysis, it is not being utilized by a member of Methanosaeta.
M. concilii is known for its high affinity to acetate, and is unable to use hydrogen for methanogenesis (Smith and Ingram-Smith, 2007). Despite this, the M. concilii genome encodes genes required for CO2 reduction, which, based on our transcriptome analysis, were all found to be highly transcriptionally active under all three conditions with the majority of the enzymes being expressed within the upper 50th percentile. Although the entire CO2 fixation branch is active, different subunits of formyl-MF dehydrogenase, the first step in CO2 reduction (Figure 4a), were expressed at variable levels across conditions. FmdA was the most highly expressed during fatty acid degradation (97th percentile), but did not appear to be expressed during hexadecane degradation (13th percentile). From previous studies, FmdA has been shown to be the catalytic subunit exhibiting amidohydrolase activity (Holm and Sander, 1997), suggesting that M. concilii may be utilizing methylamines during fatty acid degradation. Alternatively, FmdC also exhibited higher levels of expression during fatty acid degradation and is located right after the FmdA gene, so these two genes may be co-regulated. FwdG was the second most highly expressed (82nd percentile) subunit during hexadecane degradation, whereas FwdG was expressed near the median during fatty acid degradation (42nd percentile). M. concilii has previously been shown to form conductive aggregates with species of Geobacter, suggesting that direct interspecies electron transfer (DIET) can occur between these species (Morita et al., 2011). It has recently been demonstrated by the Lovley group that Methanosaeta can reduce CO2 while accepting electrons by DIET using a transcriptomic and radiotracer approach (Rotaru et al., 2013). The higher expression of FwdG during hexadecane degradation potentially indicates that a source of electrons in addition to acetate is being utilized for methanogenesis, as FwdG is predicted to be a potential electron carrier. Although we cannot directly confirm that DIET is occurring in this consortium, it is possible that Smithella is transferring electrons to M. concilii for CO2 reduction in addition to acetate during hexadecane degradation.
Methanocalculus, a member of the family Methanocorpusculaceae that can reduce CO2 with hydrogen or formate as an electron donor (Zellner et al., 1989), was the predominant methanogen under hexadecane-degrading conditions. Members of this family were previously found in methanogenic alkane-degrading communities, composing around 20–30% of the archaeal community (Grabowski et al., 2005; Gray et al., 2011). Although we could not directly confirm the activity of Methanocalculus, both formate dehydrogenases and ech hydrogenases were actively transcribed during growth with hexadecane. It has been suggested that methanogenic degradation of long-chain alkanes requires the presence of both acetoclastic as well as hydrogenotrophic methanogens (Zengler et al., 1999; Jones et al., 2008). Thus, it is possible that Methanocorpusculaceae is fulfilling this role in concurrence with M. concilii in this community.
The hydrogenotrophic M. marisnigri, a member of the Methanomicrobiales, was most abundant during hexadecane degradation (Figure 4b), but only a small percentage of reads (11 762, representing 0.02%) mapped under this condition. This discrepancy likely arises from sequence dissimilarity between sequenced strains available in databases and the actual organism present in the community. Thus, in order to fully assess the activity of specific community members, draft genomes of targeted species must be obtained by a single-cell approach used here or alternatively by a computational approach that bins metagenomic sequences (Chatterji et al., 2008; Dick et al., 2009; Albertsen et al., 2013).
Smithella, a genus that to date has not been sequenced, was targeted for this study. Integration of single-cell genome sequencing and metatranscriptomics allowed us to identify potential genes involved in anaerobic hexadecane degradation. Extension of the metatranscriptomic data sets to additional representative genomes of community members yielded insight into the nature of potential syntrophic interactions Smithella participates in. Furthermore, applying species-specific genome-scale analysis of this community across multiple conditions provided insights into the mechanisms driving major shifts in abundance and activity of key community constituents. Moving beyond purely descriptive, meta-level analyses of communities will eventually allow an understanding of the plasticity of community composition and the capacity of individual members to accommodate and respond to environmental perturbation.
Accession codes
References
Aeckersberg F, Bak F, Widdel F . (1991). Anaerobic oxidation of saturated hydrocarbons to CO2 by a new type of sulfate-reducing bacterium. Arch Microbiol 156: 5–14.
Aitken CM, Jones DM, Maguire MJ, Gray ND, Sherry A, Bowler BFJ et al (2013). Evidence that crude oil alkane activation proceeds by different mechanisms under sulfate-reducing and methanogenic conditions. GeochimCosmochim Acta 109: 162–174.
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH . (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538.
Anders S, Huber W . (2010). Differential expression analysis for sequence count data. Genome Biol 11: R106.
Aziz R, Bartels D, Best A, DeJongh M, Disz T, Edwards R et al (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9: 75.
Breuil C, Kushner DJ . (1980). Effects of lipids, fatty acids, and other detergents on bacterial utilization of hexadecane. Can J Microbiol 26: 223–231.
Callaghan AV, Morris BEL, Pereira IAC, McInerney MJ, Austin RN, Groves JT et al (2012). The genome sequence of Desulfatibacillum alkenivorans AK-01: a blueprint for anaerobic alkane oxidation. Environ Microbiol 14: 101–113.
Chatterji S, Yamazaki I, Bai Z, Eisen J . (2008). CompostBin: A DNA composition-based algorithm for binning environmental shotgun reads. in: Vingron M, Wong L (eds) Research in Computational Molecular Biology. Springer: Berlin Heidelberg, Germany, pp 17–28.
Cheng L, Ding C, Li Q, He Q, Dai L-r Zhang H . (2013). DNA-SIP reveals that Syntrophaceae play an important role in methanogenic hexadecane degradation. PLoS One 8: e66784.
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F et al (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36: W465–W469.
Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP et al (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol 10: R85.
Gieg LM, Duncan KE, Suflita JM . (2008). Bioenergy production via microbial conversion of residual oil to natural gas. Appl Environ Microbiol 74: 3022–3029.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I et al (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol 29: 644–652.
Grabowski A, Nercessian O, Fayolle F, Blanchet D, Jeanthon C . (2005). Microbial diversity in production waters of a low-temperature biodegraded oil reservoir. FEMS Microbiol Ecol 54: 427–443.
Gray ND, Sherry A, Grant RJ, Rowan AK, Hubert CRJ, Callbeck CM et al (2011). The quantitative significance of Syntrophaceae and syntrophic partnerships in methanogenic degradation of crude oil alkanes. Environ Microbiol 13: 2957–2975.
Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL . (2010). High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol 28: 977–982.
Holm L, Sander C . (1997). An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins 28: 72–82.
Jones DM, Head IM, Gray ND, Adams JJ, Rowan AK, Aitken CM et al (2008). Crude-oil biodegradation via methanogenesis in subsurface petroleum reservoirs. Nature 451: 176–180.
Langmead B, Trapnell C, Pop M, Salzberg S . (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25.
Liu T, Wang F, Guo L, Li X, Yang X, Lin A . (2012). Biodegradation of n-hexadecane by bacterial strains B1 and B2 isolated from petroleum-contaminated soil. Sci China Chem 55: 1968–1975.
Liu Y, Balkwill DL, Aldrich HC, Drake GR, Boone DR . (1999). Characterization of the anaerobic propionate-degrading syntrophs Smithella propionica gen. nov., sp. nov. and Syntrophobacter wolinii. Int J Syst Bacteriol 49: 545–556.
Maestrojuán GM, Boone DR, Xun L, Mah RA, Zhang L . (1990). Transfer of Methanogenium bourgense, Methanogenium marisnigri, Methanogenium olentangyi, and Methanogenium thermophilicum to the genus Methanoculleus gen. nov., emendation of Methanoculleus marisnigri and Methanogenium, and description of new strains of Methanoculleus bourgense and Methanoculleus marisnigri. Int J Syst Bacteriol 40: 117–122.
McInerney MJ, Rohlin L, Mouttaki H, Kim U, Krupp RS, Rios-Hernandez L et al (2007). The genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. Proc Nat Acad Sci USA 104: 7600–7605.
Morita M, Malvankar NS, Franks AE, Summers ZM, Giloteaux L, Rotaru AE et al (2011). Potential for direct interspecies electron transfer in methanogenic wastewater digester aggregates. MBio 2: e00159–e00111.
Movahedi NS, Forouzmand E, Chitsaz H . (2012). De novo co-assembly of bacterial genomes from multiple single cells. 2012 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 1–5.
Okugawa S, Moayeri M, Pomerantsev AP, Sastalla I, Crown D, Gupta PK et al (2012). Lipoprotein biosynthesis by prolipoprotein diacylglyceryl transferase is required for efficient spore germination and full virulence of Bacillus anthracis. Mol Microbiol 83: 96–109.
Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S et al (2009). Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37: e123.
Rojo F . (2009). Degradation of alkanes by bacteria. Environ Microbiol 11: 2477–2490.
Rotaru A-E, Shrestha P, Liu F, Shrestha M, Markovaite B, Shrestha D et al (2013). Elucidating electrical connections for direct interspecies electron transfer in defined syntrophic methanogenic co-cultures. Final Program American Society for Microbiology 113th General Meeting P 143.
Scherr KE, Lundaa T, Klose V, Bochmann G, Loibner AP . (2012). Changes in bacterial communities from anaerobic digesters during petroleum hydrocarbon degradation. J Biotechnol 157: 564–572.
Seth-Smith HMB, Harris SR, Skilton RJ, Radebe FM, Golparian D, Shipitsyna E et al (2013). Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res 5: 855–866.
Siddique T, Penner T, Semple K, Foght JM . (2011). Anaerobic biodegradation of longer-chain n-alkanes coupled to methane production in oil sands tailings. Environ Sci Technol 45: 5892–5899.
Shrestha PM, Rotaru AE, Summers ZM, Shrestha M, Liu F, Lovley DR . (2013). Transcriptomic and genetic analysis of direct interspecies electron transfer. Appl Environ Microbiol 79: 2397–2404.
Smith KS, Ingram-Smith C . (2007). Methanosaeta, the forgotten methanogen? Trends Microbiol 15: 150–155.
So CM, Phelps CD, Young LY . (2003). Anaerobic transformation of alkanes to fatty acids by a sulfate-reducing bacterium, strain Hxd3. Appl Environ Microbiol 69: 3892–3900.
Swan BK, Martinez-Garcia M, Preston CM, Sczyrba A, Woyke T, Lamy D et al (2011). Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science 333: 1296–1300.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ et al (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515.
Tzintzun-Camacho O, Loera O, Ramírez-Saad HC, Gutiérrez-Rojas M . (2012). Comparison of mechanisms of hexadecane uptake among pure and mixed cultures derived from a bacterial consortium. Int Biodeterioration Biodegrad 70: 1–7.
Wang L-Y, Gao C-X, Mbadinga SM, Zhou L, Liu J-F, Gu J-D et al (2011). Characterization of an alkane-degrading methanogenic enrichment culture from production water of an oil reservoir after 274 days of incubation. Int Biodeterioration Biodegrad 65: 444–450.
Wang Z, Gerstein M, Snyder M . (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63.
Wilkes H, Rabus R, Fischer T, Armstroff A, Behrends A, Widdel F . (2002). Anaerobic degradation of n-hexane in a denitrifying bacterium: Further degradation of the initial intermediate (1-methylpentyl)succinate via C-skeleton rearrangement. Arch Microbiol 177: 235–243.
Yoon HS, Price DC, Stepanauskas R, Rajah VD, Sieracki ME, Wilson WH et al (2011). Single-cell genomics reveals organismal interactions in uncultivated marine protists. Science 332: 714–717.
Zedelius J, Rabus R, Grundmann O, Werner I, Brodkorb D, Schreiber F et al (2011). Alkane degradation under anoxic conditions by a nitrate-reducing bacterium with possible involvement of the electron acceptor in substrate activation. Environ Microbiol Rep 3: 125–135.
Zellner G, Stackebrandt E, Messner P, Tindall B, De Macario EC, Kneifel H et al (1989). Methanocorpusculaceae fam. nov., represented by Methanocorpusculum parvum, Methanocorpusculum sinense spec. nov. and Methanocorpusculum bavaricum spec. nov. Arch Microbiol 151: 381–390.
Zengler K, Richnow HH, Rossello-Mora R, Michaelis W, Widdel F . (1999). Methane formation from long-chain alkanes by anaerobic microorganisms. Nature 401: 266–269.
Zengler K, Palsson BO . (2012). A road map for the development of community systems (CoSy) biology. Nat Rev Micro 10: 366–372.
Acknowledgements
We thank Rohini Patel for technical assistance and Ramunas Stepanauskas (Bigelow Laboratory for Ocean Sciences) for assistance with single-cell genome amplification. We also thank Pravin Shrestha and Derek Lovley (UMass, Amherst) for supplying total RNA of the Geobacter consortium. This work was supported by the Office of Science (BER), U.S. Department of Energy, award no. DE-SC0004485 and DE-SC0004917. This Whole-Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AWGX00000000. The version described in this paper is version AWGX01000000. Metatranscriptome analysis can be accessed through GEO, accession no. GSE49830.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Embree, M., Nagarajan, H., Movahedi, N. et al. Single-cell genome and metatranscriptome sequencing reveal metabolic interactions of an alkane-degrading methanogenic community. ISME J 8, 757–767 (2014). https://doi.org/10.1038/ismej.2013.187
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.187
Keywords
This article is cited by
-
Implementation of Genetic Engineering and Novel Omics Approaches to Enhance Bioremediation: A Focused Review
Bulletin of Environmental Contamination and Toxicology (2022)
-
Metabolic cooperation and spatiotemporal niche partitioning in a kefir microbial community
Nature Microbiology (2021)
-
Identity and hydrocarbon degradation activity of enriched microorganisms from natural oil and asphalt seeps in the Kurdistan Region of Iraq (KRI)
Biodegradation (2021)
-
Bacterial and archaeal diversity in oil fields and reservoirs and their potential role in hydrocarbon recovery and bioprospecting
Environmental Science and Pollution Research (2021)
-
Methanogenic biodegradation of C9 to C12n-alkanes initiated by Smithella via fumarate addition mechanism
AMB Express (2020)

{kind=link}
{kind=link}
{kind=link}