
Abstract
Disruption of the microbiome often correlates with the appearance of disease symptoms in metaorganisms such as corals. In Black Band Disease (BBD), a polymicrobial disease consortium dominated by the filamentous cyanobacterium Roseofilum reptotaenium displaces members of the epibiotic microbiome. We examined both normal surface microbiomes and BBD consortia on Caribbean corals and found that the microbiomes of healthy corals were dominated by Gammaproteobacteria, in particular Halomonas spp., and were remarkably stable across spatial and temporal scales. In contrast, the microbial community structure in black band consortia was more variable and more diverse. Nevertheless, deep sequencing revealed that members of the disease consortium were present in every sampled surface microbiome of Montastraea, Orbicella and Pseudodiploria corals, regardless of the health status. Within the BBD consortium, we identified lyngbic acid, a cyanobacterial secondary metabolite. It strongly inhibited quorum sensing (QS) in the Vibrio harveyi QS reporters. The effects of lyngbic acid on the QS reporters depended on the presence of the CAI-1 receptor CqsS. Lyngbic acid inhibited luminescence in native coral Vibrio spp. that also possess the CAI-1-mediated QS. The effects of this naturally occurring QS inhibitor on bacterial regulatory networks potentially contribute to the structuring of the interactions within BBD consortia.
Similar content being viewed by others


Introduction
Corals are holobionts, complex metaorganisms that consist of the invertebrate animal host, dinoflagellate endosymbionts and microbial communities associated with them. Coral microbiota have important roles in the health of the holobiont and contribute importantly to nutrient acquisition (Bourne et al., 2009; Bosch and McFall-Ngai, 2011). Displacement of primary commensals with other members of the microbiome or shifts in the abundance of microbiome members have been linked with the appearance of disease signs and polymicrobial diseases (Cardenas et al., 2012; Vezzulli et al., 2013; Roder et al., 2014b).
Black Band Disease (BBD) is the first reported coral disease and the most widely distributed polymicrobial disease of corals, affecting at least 40 species worldwide (Edmunds, 1991; Littler and Littler, 1996; Sussman et al., 2006; Montano et al., 2013). The disease is recognized by the appearance of a dense, very dark-purple (black) mat that is the visible accumulation of filamentous cyanobacteria. BBD migrates across the surface of the coral at a rate of up to 1 cm per day (Richardson, 1998), leaving behind bare coral skeleton. Microscopic examination has shown that both thick cyanobacterial filaments and smaller unidentified filaments are present in the leading edge of the disease front (Miller et al., 2011). Similar to other cyanobacterial mats, the black band consortium is stratified, with increasing levels of anoxia and sulfide with depth (Glas et al., 2012), effectively smothering and poisoning the coral tissue beneath.
Meta-analysis of published clone library studies of BBD microbial communities revealed that, with few exceptions, microbial species composition of BBD communities did not correlate with the species of the host coral (Miller and Richardson, 2011). Nevertheless, the same cyanobacterial Operational Taxonomic Unit (OTU) (now classified as Roseofilum reptotaenium; Frias-Lopez et al., 2003; Casamatta et al., 2012) was present in over 70% of the samples (Miller and Richardson, 2011), indicating that this organism is critical for the appearance or progression of BBD. Similar to other filamentous cyanobacteria, Roseofilum can be cultured in unialgal, but not axenic cultures; therefore, it can only be confirmed as a pathogen of BBD with modified Koch’s postulates, that is, it is part of a polymicrobial consortium and cannot be confirmed as the primary pathogen (Casamatta et al., 2012). In addition to R. reptotaenium, three OTUs (Bacteroidetes and Alphaproteobacteria) were present in 13% of the samples, with other OTUs found in <7% of the samples (Miller and Richardson, 2011). Even though sulfate reduction was shown to be critical to the progression of BBD (Glas et al., 2012), Desulfovibrio profundus (which is thought to be responsible for the H2S production) was detected in only ~5% of the clone libraries analyzed (Miller and Richardson, 2011). These discrepancies could be due to technical issues (e.g., amplification biases, low coverage of the microbiome in clonal libraries), seasonal and/or regional differences in the BBD composition or function-based (rather than taxonomy-based) assembly of the BBD community. Therefore, there is a need for a comprehensive characterization of BBD microbiota across multiple scales. Furthermore, to better understand initiation of BBD, its progression over a coral colony and ecosystem-wide spread, it is important to characterize the composition of the epimicrobiota of asymptomatic corals across temporal and spatial scales within ecosystems where BBD is known (or not known) to occur. These were some of the objectives of this study.
How BBD is initiated and transmitted remains uncertain. It spikes in warmer, sunnier months, and once a BBD outbreak takes place within a given ecosystem, it tends to reoccur each year (Zvuloni et al., 2009; Sato et al., 2009; Kuehl et al., 2011; Muller and van Woesik, 2011; Sato et al., 2011). Individual coral colonies in which BBD has halted are 3.5 times more likely to develop signs of the disease in subsequent years (Sato et al., 2009). Analyses of the within-ecosystem, spatiotemporal patterns of the appearance of BBD point to various models, including transmission by water currents, stochastic appearance of lesions and direct colony-to-colony transmission (Bruckner et al., 1997; Sato et al., 2009; Zvuloni et al., 2009). These observations also do not rule out a hypothesis that the spatiotemporal distribution of BBD cases on the reef correlates with water quality parameters (Page and Willis, 2006), which may serve as environmental triggers for community shifts mediated by BBD members. Alternate hypotheses, including vector-borne transmission from non-coral reservoirs or between diseased and healthy corals have also been suggested (Aeby and Santavy, 2006; Casey et al., 2014). Cyanobacteria similar to those associated with BBD, including the genera Leptolyngbya, Geitlerinema, Oscillatoria and Phormidium, were found in microbiomes associated with turf, crustose coralline and macroalgae, potentially implicating these organisms as alternate hosts or reservoirs of the pathogens (Barott et al., 2011).
Microbe–microbe interactions within BBD remain only partially understood. It is clear that complex transitions within coral microbiota are a prelude to the development of BBD (Sato et al., 2010). Changes in the biogeochemical conditions (gradients of H2S, pH, oxygen and light availability) within the BBD mat contribute to the stratification of microorganisms (Sato et al., 2011; Glas et al., 2012), although mechanisms of cell-to-cell interactions driving the structuring of BBD consortia remain unknown. Recent discovery of N-acyl-homoserine lactones (AHLs) and AI-2 quorum sensing (QS) signals within BBD (Zimmer et al., 2014) indicates that QS is likely involved in the structuring or functionality of BBD. QS is a mechanism of bacterial population density-dependent gene regulation, which has been shown to contribute importantly to structuring of host-associated microbiota (Miller and Bassler, 2001) and to interactions within microbial communities (Ng and Bassler, 2009). Bacteria use QS to express virulence genes, spread over surfaces, form and disperse biofilms and produce ‘public goods’ that can be used by the entire microbial community. However, it is not known how QS and signal interference contribute to coral diseases in general and to BBD in particular.
Here, we documented differences in the composition of microbiomes of healthy corals and those afflicted with BBD in distinct Caribbean ecosystems. To better understand interactions within BBD, secondary metabolites were extracted from the mat. We tested the hypothesis that an abundant cyanobacterial compound, lyngbic acid, isolated from BBD, interfered with QS, a signal perception and regulatory mechanism involved in structuring of microbial communities and their functions.
Materials and methods
Sample collection
Samples of BBD consortia and surface mucus from the Florida Keys, Belize and Honduras were collected by aspiration with needle-less sterile syringes that were uncapped immediately before the sample collection. A total of 60 samples were collected, including 30 BBD consortia, 20 surface mucus layer samples overlying healthy tissue on corals with BBD and 10 mucus samples from asymptomatic corals (Supplementary Table S1). Some of the corals in Belize were tagged so that the same coral was sampled at up to three time points, although not all tagged corals were resampled at every time point because of difficulties in locating tags on some of the subsequent samplings. Surface mucus from healthy tissue on corals with BBD was collected at least 10–15 cm away from an active lesion. Samples were spun in a microfuge at 4000 r.p.m. for 5 min, seawater was decanted and the pelleted mucus was preserved with ~5–10 volumes of RNAlater (Qiagen, Germantown, MD, USA) and stored at −20 °C until extraction of nucleic acids. Seawater was collected by boat off Carrie Bow Cay, Belize from an area adjacent to but not overlying the reef and transported to shore. Seawater was filtered with a 0.2-μm filter in duplicate, with a total of 3 L of seawater for each filter. The filters were placed in a 15-ml conical tube, immersed in RNAlater and stored −20 °C until extraction of nucleic acids.
Coral-associated vibrios were isolated in April 2014 from healthy mucus and from the black band layer of Montastraea cavernosa from Florida on marine agar or TCBS (thiosulfate-citrate-bile salts-sucrose) agar. Individual colonies were sequentially streaked to isolation a minimum of three times and identified through sequencing of the V6-V8 region of the 16S rRNA gene with primers F968 and R1401 (Nübel et al., 1996). A total of 29 coral-associated vibrios were screened for the production of CAI-1 autoinducer in AB medium (Supplementary Table S4). Of them, two bioluminescent isolates Vibrio spp. BBD33 (from black band) and BBD69 (from healthy mucus) were selected for subsequent assays with lyngbic acid.
QS assays
Cultures of V. harveyi and Escherichia coli reporters (Supplementary Table S2) were grown overnight with antibiotics in AB medium (for V. harveyi; Bassler et al., 1994) or in LB (for E. coli) with kanamycin (50 μg ml−1), chloramphenicol (10 μg ml−1) or ampicillin (200 μg ml−1) as appropriate to the genotype. To determine whether coral vibrios make CAI-1 signal, overnight AB cultures were spun, filtered through a 0.2 micron filter and mixed (at ~30%) with a diluted culture of JMH626 (ΔluxN luxQ::Tn5 cqsA::Cm), which only responds to CAI-1 (Supplementary Table S2 and Henke and Bassler, 2004). For the bioassays with lyngbic acid, a panel of the V. harveyi QS reporters and Vibrio spp. BBD33 and BBD69 were grown overnight, diluted at least 5000-fold, essentially as in Henke and Bassler (2004) and then added to the test wells containing fivefold serial dilutions of lyngbic acid. Dilutions of lyngbic acid were made in ethanol, and before adding reporter suspensions, the solvent was completely evaporated in a laminar flow hood. Control wells contained residue from the dried carrier solvent. Suspensions of the reporters were mixed with the dried lyngbic acid residue in each well by vigorous pipetting. Luminescence of the reporters was quantified over time with a Victor-2 multimode microtiter plate reader (Perkin-Elmer, Waltham, MA, USA).
‘Indirect’ assays were conducted to determine whether the addition of the cognate CAI-1 signal overrides the effect of lyngbic acid. Serial dilutions of lyngbic acid were prepared as above. Thirty microliters of filter-sterilized culture filtrates of V. harveyi KM413 (ΔluxS ΔluxM), which produces only the CAI-1 signal (Henke and Bassler, 2004) were added to all wells with lyngbic acid and then mixed with 90μl of the diluted suspension of V. harveyi JMH626 (which only responds to CAI-1) (Supplementary Table S2). Luminescence was measured every 30 min using the multimode microtiter plate reader.
16S Illumina tag sequencing
Genomic DNA for 16S Illumina tag sequencing was extracted with a PowerSoil DNA Isolation Kit (MoBio, Carlsbad, CA, USA). The V6 region of 16S rRNA genes was amplified in triplicate for each sample with previously reported Illumina-compatible primers designed for bacteria (Eren et al., 2013). Samples were amplified in 25 μl reactions containing 0.5 U Phusion High-Fidelty Polymerase (New England Biolabs, Ipswich, MA, USA), 1 × Phusion HF Reaction Buffer, 0.75 μl dimethyl sulfoxide and 0.2 mm each dNTP. Triplicate PCR amplifications were pooled for each sample, cleaned with a MinElute Kit (Qiagen, Germantown, MD, USA), visualized on an ethidium bromide stained 1% agarose gel and quantified by NanoDrop (ThermoScientific, NanoDrop Products, Wilmington, DE, USA). Two hundred nanograms of each cleaned amplicon library was submitted to the Genomics Core Facility at Pennsylvania State University where the pooled libraries were size selected with a 2% agarose PippinPrep cassette to produce a narrow range of fragment sizes from 200 to 240 bp for sequencing (confirmed by a bioanalyzer) and cleaned again to remove agarose. Sequencing was performed on an Illumina MiSeq with a 150-bp paired-end protocol, using single indexing.
Sequencing reads were parsed by Illumina index at the sequencing center. Reads were then further parsed by the inline barcode, paired reads merged and primers and adaptors removed using a combination of tools in cutadapt (Martin 2011), Galaxy (Giardine et al., 2005; Blankenberg et al., 2010; Goecks et al., 2010) and eautils (Aronesty, 2011). Parsed raw sequencing reads are publicly available through NCBI’s Sequence Read Archive under the BioProject ID PRJNA269585. Sample names were added to the definition lines of sequencing reads using sed and concatenated into one fasta file, to make them compatible for analysis in QIIME v.1.8 (Caporaso et al., 2010). Clustering of OTUs at 97% similarity was performed with the subsampled open-reference OTU picking method (Rideout et al., 2014), with no removal of singletons. The Greengenes reference data set version 13.8 (DeSantis et al., 2006) was used as the reference for OTU picking and for taxonomy assignment with uclust (Edgar, 2010). OTUs that were not assigned taxonomy were manually queried with BLASTn (Altschul et al., 1990) against the non-redundant nucleotide collection of the NCBI (Mizrachi, 2002). OTUs identified as mitochondrial DNA or as chloroplasts were removed from further analyses. Community structure was analyzed in R with phyloseq (McMurdie and Holmes, 2013) and plotted with ggplot2 (Wickham, 2009). Analysis of similarities (ANOSIM) was performed in R using VEGAN v.2.0-8 (Dixon, 2003). Network analysis of interactions between genera was conducted on a subset of 52 of the 60 coral microbiomes, focusing on the Florida and Belize locations that had both healthy and diseased samples from Orbicella and Montastraea hosts. Significant positive and negative correlations between genera with a minimum occurrence of 10 (out of 52 microbiomes) were determined by support of four separate measures: Pearson's correlation, Spearman's correlation, Bray–Curtis dissimilarity or Kullback–Leibler dissimilarity, using the CoNet app (Faust et al., 2012) in Cytoscape v.3.0.2 (Shannon et al., 2003). Networks from the four measures were merged by intersection, keeping only interactions with support from all methods. Rows were shuffled and the measures were performed with 1000 iterations, Benjamini–Hochberg multiple test correction was applied and highly interconnected regions (clusters) were identified with MCODE (Bader and Hogue, 2003).
Isolation of lyngbic acid
Lyngbic acid was isolated and completely characterized from two separate collections of BBD, one from the Florida Keys and one from Belize. The cyanobacterial mat from the black band consortium on M. cavernosa corals on the Looe Key Reef, Florida was collected on 23 April 2014. The sample was lyophilized to give a dry weight of 3.672 g and then extracted two times with 50 ml of MeOH. The extract was concentrated by rotary evaporation at 45 °C under reduced pressure to give 1.03 g of the MeOH-soluble extract. This extract was partitioned between ethyl acetate (EtOAc) and H2O, and the EtOAc-soluble fraction was rotary evaporated. The EtOAc-soluble fraction (0.105 g) was chromatographed on a C18 column using an MeOH:H2O step gradient system to give six subfractions. A portion (0.003 g) of subfraction 4 (0.012 g), eluted with MeOH:H2O (4:1) was further separated by reversed-phase HPLC (semi-prep, 5 μm, RP-C18) using MeOH:H2O (19:1) to give 1.2 mg of lyngbic acid (yield 0.13% dry wt, 4.8% of EtOAc-soluble fraction). Similarly, cyanobacterial mats from the BBD zones of coral species of Orbicella and Pseudodiplora in Belize were collected on July 13-17, 2013. The combined freeze-dried material (9.43 g) was extracted with MeOH saturated with helium gas. This MeOH extract (1.82 g) was partitioned between EtOAc and H2O to give the EtOAc-soluble fraction (0.237 g) and H2O-soluble fraction. The EtOAc extract was chromatographed on a C18 column (16 g) using MeOH−H2O step gradient system to give six subfractions. A portion (0.004 g) of the subfraction 4 (0.023 g), eluted with MeOH-H2O (4:1), was further separated by reversed-phase HPLC using the same conditions to give 0.9 mg of lyngbic acid (yield 0.10% dry wt, 4.1% of EtOAc-soluble fraction). The structure of lyngbic acid was determined using the proton nuclear magnetic resonance spectroscopy, high-resolution mass spectrometry and optical rotation data and confirmed by comparison with published data and data from our previously isolated samples of lyngbic acid (Cardellina et al., 1978; Kwan et al., 2010; Soares et al., 2015). The presence of lyngbic acid in other collections of BBD (Belize: 1 March 2013; FL Keys: Looe Key 17 May 2013, 12 September 2013, 20 November 2013; and Wonderland Reef 10 June 2013) was determined by identifying the presence of the key methoxy and unsaturation proton signals in the nuclear magnetic resonance spectroscopy spectra of the extracts and fractions.
Results
Community structure of healthy and BBD coral microbiomes
The bacterial community structure and diversity of both healthy corals and those with BBD was determined for 60 coral surface microbiomes from Carrie Bow Cay, Belize, Looe Key in the Florida Keys, and four locations in Honduras, as well as for two background seawater samples from Carrie Bow Cay, Belize. A total of 14 571 218 bacterial small subunit rRNA gene sequences were analyzed after quality filtering, with 7497 to 750 674 sequences per sample (Supplementary Table S1). Distinct bacterial communities were detected in the surface microbiomes of healthy corals (epibiomes), in seawater and in the BBD consortia (Figure 1). Epibiomes were remarkably conserved and consisted primarily of Gammaproteobacteria (in particular, the genera Halomonas and Moritella) and Renibacterium (Figure 2). Epibiomes exhibited the lowest OTU richness and α-diversity compared with bacterial communities in seawater and BBD (Figure 3). Overall, the polymicrobial BBD consortia displayed more variable bacterial community structure than epibiomes (e.g., Figure 1 and Supplementary Figure S1), but this variability did not correspond with sample location (ANOSIM R statistic=0.0867, significance from permutations=0.044) (Figure 1a) or with coral species (ANOSIM R statistic=0.1456, significance from permutations=0.014) (Figure 1b), where an ANOSIM R statistic near zero indicates random variation between groups. However, a hypothesis that the composition of BBD varied seasonally was supported. BBD communities collected in Belize in cooler times of the year when the disease is thought to be less active were distinct from the BBD microbial communities sampled in warm times of the year when the disease was active (Figure 1). Furthermore, winter BBD samples tended to cluster with the seawater samples (Figure 1). Seawater samples exhibited the highest OTU richness and α-diversity (Figure 3).

Comparison of bacterial community structure in healthy and diseased coral microbiomes. Bacterial community similarity based on Morisita–Horn β-diversity of Illumina MiSeq 16S rRNA gene libraries in the surface microbiomes of corals with and without BBD, in comparison with overlying seawater, with health state indicated by color and collection location (a) or coral host (b) indicated by symbol shape. Black Band communities sampled in February were consistently different from summer Black Band communities. Healthy surface microbiomes clustered together regardless of collection location or coral species.

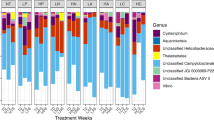
Distribution of abundant bacterial genera in healthy and diseased coral microbiomes. Box and whisker plot of the relative proportion of sequencing reads assigned to 35 most abundant bacterial taxa of surface microbiomes from corals with BBD (n=23), with BBD in winter (n=7), in healthy tissue on corals with BBD (n=20), in healthy coral heads (n=10), and in comparison with seawater above the reef (n=2). Points outside of the box and whiskers represent data outliers. Taxa were classified to genus when possible; taxa labeled with higher taxonomic classifications did not match named lineages within the database. Families labeled ‘(A)’ or ‘(B)’ represent separate lineages, which were not classified to more specific levels and match taxa labels in Figure 4 and Supplementary Figure S1.

α-Diversity in healthy and diseased coral microbiomes. Comparison of observed taxonomic richness (number of OTUs clustered at 97% similarity), and the Chao1 (with s.e.), ACE and Shannon diversity indices in ‘B’: summer BBD consortia; ‘W’: winter BBD consortia; ‘H’: healthy surface microbiomes; and ‘S’: seawater. The background seawater samples were most diverse, and diseased microbiomes tended to be more diverse comapred with healthy microbiomes.
Overall, bacterial communities in healthy mucus had high proportions of Halomonas, whereas black band consortia had high proportions of Roseofilum (Figure 2 and Supplementary Figure S1). The filamentous, nonheterocystous, phycoerythrin-rich cyanobacterium Roseofilum reptotaenium was detected in every BBD sample, although great variation in its abundance was observed (Figure 2 and Supplementary Figure S2). Although the dominant cyanobacterial OTUs in the Illumina libraries were classified as ‘Leptolyngbya’ by Greengenes v.13.8, comparison of full-length cyanobacterial 16S rRNA genes recovered from Belize and Florida Keys metagenomic libraries prepared from the same DNA extractions and from a Roseofilum enrichment culture (KP689103) (Meyer et al., in preparation) were >99% similar to sequences from the uncultured cyanobacterium (AY038527) identified in the first molecular characterization of Caribbean BBD mats (Frias-Lopez et al., 2002), the cultured BBD cyanobacterium Roseofilum reptotaenium strain 101-1 (EU743965) from Caribbean Pseudodiploria strigosa (Casamatta et al., 2012), and Pseudoscillatoria coralii (FJ210722) from Favia corals in the Red Sea (Rasoulouniriana et al., 2009). Collectively, these results support the hypothesis that R. reptotaenium is the main cyanobacterium present in BBD mats.
In addition to Roseofilum, the disease consortia were dominated by uncultured genera of Rhodobacteraceae and Bacteroidales, Fusibacter, and Desulfovibrio (Figure 2). The dominant OTUs classified as uncultured Rhodobacteraceae closely match (⩽2 nucleotide differences along the 60-bp length of the V6 amplicon sequence) uncultured Alphaproteobacteria recovered from Siderastrea corals with BBD (DQ446102) (Sekar et al., 2006), from healthy Orbicella annularis corals (FJ952692) (Rypien et al., 2010), and from corals with Porites White Patch Syndrome (KF179798) (Sere et al., 2013). The dominant OTUs classified as uncultured Bacteroidales were not closely related to any Bacteroidetes detected in coral microbiomes.
Healthy coral microbiota are stable over time, depth and location
To test the hypothesis that the epibiome of healthy corals is stable over spatial and temporal gradients, we sampled healthy surface microbial communities from M. cavernosa, O. annularis, O. faveolata and P. strigosa in Belize and Florida Keys over 2 years in February, April, May, June, July and August. Epibiomes generally clustered together (Supplementary Figure S3). Healthy microbiomes from O. annularis collected over a range of depths were indistinguishable from those collected in shallow waters (Supplementary Figure S3C). Importantly, all samples (including those collected from depths ranging from ~12 to 21 m, where BBD is not known to occur) contained R. reptotaenium, the cyanobacterium associated with BBD. Up to 0.3% of sequencing reads from healthy corals were identified as Roseofilum. These observations clearly establish that coral epibiomes are stable across temporal and spatial gradients. Furthermore, these observations support the hypothesis that R. reptotaenium is a normal, albeit minor, member of the coral microbiota, even at depths where BBD does not occur.
Microbiome shifts and BBD progression
In addition to the overall characterization of healthy and black band microbiomes, we examined spatial patterns in the distribution and progression of BBD. First, we fortuitously located adjacent colonies of O. annularis and P. strigosa, which both displayed signs of BBD (Supplementary Figure S4). A comparison of their microbiomes revealed that the bacterial communities from healthy tissues were conserved and clustered together, whereas BBD communities on adjacent corals were distinct (Supplementary Figure S4). Second, examination of the surface microbiome at 5 cm increments away from the BBD front in three separate corals of M. cavernosa showed a rapid transition from the BBD consortium, dominated by R. reptotaenium, to the conserved epibiome consisting primarily of Gammaproteobacteria, in particular, the genera Halomonas, Moritella and Marinobacter (Supplementary Figure S5). It is important to note that in the asymptomatic transition zone (5 cm away from the lesion front), R. reptotaenium was significantly represented (up to 4.5% of the microbiome), and the cyanobacterium was present as a minor (⩽0.1%) component of the stable microbiomes 10 and 15 cm away from the lesion front.
Interactions between healthy and BBD coral microbiota
As the black band layer migrates across the surface of infected corals, Roseofilum and its associated heterotrophs continually invade the adjacent healthy surface mucus layer. Our deep-sequencing results show that Roseofilum and other members of the polymicrobial disease consortia are present at low levels in healthy tissues and uninfected corals and that 63% of genera were shared between the two sample types. Taken together, this suggests that interactions between healthy microbiota and BBD consortia members may ultimately determine the overall health of the coral host. To test this hypothesis, we examined co-occurrence patterns of bacterial genera to identify microbe–microbe interactions that potentially influence the progression of BBD. A total of 97 significant interactions (multiple corrected P-value <0.05) were detected between 23 bacterial genera in a matched subset of Montastraea and Orbicella corals from Belize and Florida, containing 23 black band consortia and 29 healthy samples (Figure 4a). All 23 genera with non-random co-occurrence patterns were detected in both healthy and diseased samples. Overall, dominant genera within either healthy or diseased microbiomes had significant positive interactions (co-occurrences) with other microbiome members of the same health state and negative interactions (mutual exclusions) with dominant microbiome members of the opposite health state. Halomonas had a total of 17 significant interactions, including negative interactions with all of the most abundant members of the BBD consortium: Roseofilum, uncultured Rhodobacteraceae, uncultured Bacteroidales, Fusibacter and Desulfovibrio. Halomonas also had significant positive interactions with six genera, four of which were other Gammaproteobacteria, as well as the single-celled cyanobacterium Synechococcus and the actinobacterium Renibacterium. In contrast, Roseofilum had six significant negative interactions, including Halomonas and Moritella, the two most abundant genera detected in the healthy microbiomes and only one positive interaction, with Desulfovibrio, a sulfate-reducing Deltaproteobacterium. The fewer significant interactions between Roseofilum and other genera may reflect the more variable community composition in black band consortia. Further analysis of the interaction networks revealed two large clusters of highly interconnected regions of interactions. Cluster 1 involved 22 of the 23 genera with significant interactions in Figure 4a (all except Marinobacter) and revealed that many of the dominant Gammaproteobacteria, including Halomonas, Moritella, Vibrio, uncultured Enterobacteriaceae and Oceanospirillum, are hubs of connections, interacting both positively and negatively with many other taxa (Figure 4b and Supplementary Figure S6). Cluster 2 consisted of bacteria that are over-represented in BBD samples (Roseofilum, Thalassobius, Vibrio spp., uncultured Rhodobacteraceae and Fusibacter) (Figure 4c). Notably, while Rosefilum had only seven significant interactions, it was part of both clusters and several of its significant interactions were with taxa that are highly interconnected hubs (such as Vibrio spp.).

Network analysis of interactions between abundant bacterial genera. Significant interactions between bacterial genera in 52 Orbicella and Montastraea corals from Belize and Florida with and without BBD. Solid lines indicate negative interactions (mutual exclusions) and dotted lines indicate positive interactions (co-occurrences). The size of the nodes reflects the relative abundance of the genus in the entire data set, and the nodes are sorted and colored by phylum. (a) All 97 significant interactions, involving 23 genera, sorted by class and densely interconnected regions, Cluster 1 (b) and Cluster 2 (c).
Lyngbic acid is abundant within BBD
To better understand interactions within the BBD consortium, secondary metabolites present within the extracts of the BBD mats were purified. Lyngbic acid was identified as one of the major secondary metabolites in BBD extracts and represented 0.13% (dry wt) from the Florida sample and 0.10% (dry wt) from the Belize sample, and over 4% of all hydrophobic substances (which includes many primary metabolites such as pigments and lipids) extractable into ethyl acetate. It was identified based on the HRMS data (HRESI/APCIMS m/z 257.2127 [M+H]+ (calculated for C15H29O3, 257.2117)) and was further confirmed by comparing the 1H nuclear magnetic resonance spectroscopy data with an authentic sample of lyngbic acid previously isolated from other benthic filamentous marine cyanobacteria. Lyngbic acid extracted from BBD gave a specific rotation value of [α]25D −12.0 (c 0.06, CHCl3; Supplementary Figure S7), similar to the specific rotation value of lyngbic acid reported earlier from other cyanobacteria [α]26D −9 (c 7.3, CHCl3) (Cardellina et al., 1978); [α]20D −12.6 (c 0.8, CH3OH) (Kwan et al., 2010). When BBD mats were collected in quantities that were suitable for chemical extractions and subsequent analyses, the presence of lyngbic acid in BBD samples was ascertained by nuclear magnetic resonance spectroscopy and/or HRMS. It was detected in all seven tested Caribbean BBD samples, including those collected from Carrie Bow Cay in Belize and from Wonderland and Looe Keys in Florida (Supplementary Table S2). Lyngbic acid did not have a strong effect on growth of culturable coral-associated bacteria, such as Halomonas spp. and Vibrio spp. (data not shown). Therefore, it is unlikely to function as a general biocide.
Lyngbic acid inhibits CqsS-mediated QS in Vibrio spp.
With the subsequent experiments we tested the hypothesis that lyngbic acid interferes with QS in Vibrio spp. The rationale for this hypothesis was based on the observation that R. reptotaenium and Vibrio spp. co-occurred in the BBD consortia and were predicted to have a direct negative interaction (Figure 4 and Supplementary Figure S6). Vibrio spp. form highly connected hubs in the cluster representing stable microbiome (Supplementary Figure S6). Cross-species manipulation of QS is a well-characterized mechanism of microbe–microbe interactions with community-wide consequences (Dobretsov et al., 2009; Teplitski et al., 2011). Therefore, lyngbic acid extracted from BBD was subjected to a series of bioassays to elucidate its effect on QS in Vibrio spp. A suite of V. harveyi mutants, each lacking either synthases or receptors of its three QS regulatory cascades, were exposed to serial dilutions of lyngbic acid. Despite deletions in QS genes, these mutants are still luminescent, although they produce light at levels lower than those of the wild type. If lyngbic acid acted as a signal agonist, then luminescence of the strains lacking either of the QS signal synthase genes (luxM, cqsA and luxS, which are involved in synthesis of AHL, hydroxyketone CAI-1 and furanosyl borate diester AI-2 QS signals, respectively) would have increased. Instead, strong inhibition of luminescence in these mutants was observed (Figure 5 and Supplementary Table S3). Lyngbic acid did not inhibit luminescence of a constitutively luminescent E. coli DH5α pTIM2442 (Alagely et al., 2011b), indicating that it does not function as a generic toxin or a nonspecific inhibitor of luminescence at the tested concentrations (Supplementary Figure S8). To determine exactly where lyngbic acid affects QS cascades in V. harveyi, its effects on luminescence of mutants in luxN, cqsS and luxPQ (encoding receptors of AHLs, CAI-1 and AI-2 signals, respectively) were tested. Absence of a strong response to lyngbic acid in a particular receptor mutant indicates that the corresponding missing protein is critical to the signal transduction. The mutant lacking the cqsS receptor gene was the least responsive to lyngbic acid (Figure 5 and Supplementary Table S3), whereas strains in which cqsS was intact remained responsive to the cyanobacterial compound. A deletion of luxU, which functions as an ‘integration point’ for the input of all three QS signaling cascades, also strongly reduced sensitivity to lyngbic acid, consistent with the model in which effects of lyngbic acid require a QS receptor positioned below LuxU in the QS cascade (such as CqsS). Collectively, these observations suggest that the effect of lyngbic acid on QS in V. harveyi is likely mediated by the CAI-1 QS signal receptor CqsS, and lyngbic acid may function as a natural antagonist of the CqsS/CAI-1 QS system.

Effect of lyngbic acid on QS pathways. Luminescence assays were performed in seven mutant strains and one wild-type strain of Vibrio with concentrations up to 2 μm lyngbic acid to determine its effect on several different pathways in the Vibrio model. luxM, cqsA and luxS encode synthases of the AHL, furanosyl borate diester AI-2 and hydroxyketone CAI-1 signals; luxN, cqsS and luxPQ encode receptors of these signals. LuxU is a phosphorelay protein through which inputs of all three QS systems are integrated into downstream regulatory cascades. Assays were performed in duplicates in three independent experiments. Heatmaps were constructed using data from a representative experiment after 8–10 h incubation with lyngbic acid.
To more robustly test the hypothesis that lyngbic acid interferes with CqsS-mediated QS in V. harveyi, its ability to inhibit perception of the CAI-1 signal was determined using ‘indirect assays’. V. harveyi JMH626, which only contains a functional CqsS receptor (and lacks genes encoding receptors of the AHL or AI-2 signals, luxN and luxQ, as well as the CAI-1 synthase gene cqsA) (Henke and Bassler, 2004), was used as a reporter. V. harveyi JMH626 is normally dark, unless it is exposed to CAI-1 (Henke and Bassler, 2004). CAI-1 was provided in the cell-free culture filtrates of V. harveyi KM413, which lacks AHL and AI-2 synthases luxM and luxS, respectively, but has a functional Cqs QS system (Henke and Bassler, 2004). Dilutions of lynbic acid were dissolved in the 1:3 mix of the CAI-1-containing culture filtrate of KM413, and the suspension of the CAI-1-specific reporter JMH626. This ratio of the culture filtrate to the reporter was shown to elicit maximum responses (Henke and Bassler, 2004). Increasing concentrations of lyngbic acid inhibited stimulatory effects of CAI-1 (Figure 6), consistent with the postulated function of lyngbic acid as a naturally occurring inhibitor of the Cqs-mediated QS.

Competition between lyngbic acid and CAI-1. Luminescence of V. harveyi JMH626 (luxN luxQ cqsA, which does not respond to the AHL or AI-2 signals, does not make its own CAI-1 signal and only luminesces in response to the exogenous CAI-1) was measured with microtiter plate reader Victor-2 (Perkin-Elmer). Serial dilutions of lyngbic acid (indicated on x axis) were mixed with 30 μl of filter-sterilized culture filtrates of V. harveyi KM413 (ΔluxS ΔluxM, which produces only the CAI-1 signal) before adding 90 μl of the diluted suspension of JMH626. Inhibition of luminescence is indicated as the percentage of full activation of the JMH626 reporter with culture filtrates from KM413.
CAI-mediated signaling is common in coral vibrios
In addition to establishing that lyngbic acid can antagonize QS in a well-characterized reporter system, it was important to determine whether such interactions are possible in situ. Therefore, we tested the hypothesis that lyngbic acid interferes with QS in native coral-associated Vibrio spp. in a two-step experiment on vibrios isolated from both BBD mats and from healthy surface mucus layers. First, culture filtrates of coral Vibrio isolates were surveyed for their ability to induce luminescence in V. harveyi JMH626. Extracts of 27 out of 29 coral vibrios stimulated luminescence of this CqsS reporter by ~10–10 000-fold (Supplementary Table S4), indicating that CAI-mediated signaling is highly prevalent in coral vibrios. The observed differences in the levels of luminescence induced by culture filtrates are consistent with similar surveys in other Vibrio spp. (Ng et al., 2011), and may be a consequence of different amounts or structures of CAI-1-like signals produced by the isolates.
Lyngbic acid inhibits luminescence in coral vibrios
To determine whether lyngbic acid impacts native Vibrio spp., two bioluminescent strains of coral vibrios that were shown to possess the CAI-1-mediated signaling were exposed to dilution series of lyngbic acid. Similarly to the observation with the V. harveyi reporters, lyngbic acid inhibited bioluminescence of coral vibrios (Supplementary Table S3).
Discussion
Coral diseases, such as BBD, have been devastating reef ecosystems worldwide for decades (Antonius, 1973; Bourne et al., 2009; Rosenberg, 2002). Even though disease signs and ecosystem-wide consequences have been cataloged for over a dozen coral diseases, much remains to be learned about the initiation of coral diseases, as well as interactions between native microbiota and disease consortia. More generally, to what extent host-associated microbiota are involved in diseases also remains an open question, even in the best-characterized model systems.
This study focused on the characterization of BBD microbiomes and coral epibiomes in distinct ecosystems throughout the Caribbean. Healthy epibiomes were characterized by a relatively low microbial diversity and were dominated by Halomonas spp., with Moritella spp., Renibacterium spp., Kiloniella spp. also prominent in healthy epibiomes. Dominance of Halomonas spp. in commensal coral microbiomes is consistent with observations in some other corals (McKew et al., 2012); however, the closely related genus Endozoicomonas tends to be dominant in others (Morrow et al., 2012; Bayer et al., 2013; Meyer et al., 2014).
BBD microbiomes were more diverse than healthy coral epibiomes; similarly, an increase in bacterial diversity was characteristic of other coral diseases (Sunagawa et al., 2009; Closek et al., 2014; Roder et al., 2014a). Even though α-diversity of BBD microbiomes was generally higher compared with that in coral commensal epibiomes, the same key members (R. reptotaeneum, Bacteroidales, Rhodobacteriaceae, Fusibacter spp. and Desulfovibrio spp.) were the most prominent BBD members in the samples from the Florida Keys, Belize and three locations in Honduras. This suggests that the assembly of the BBD consortia likely involves a combination of environmental factors and interactions within the microbiomes. Interestingly, there were seasonal differences in the BBD microbiome composition. BBD communities in winter had a significantly higher representation of Rhodobacteraceae, and a relative decrease in abundance of R. reptotaenium. This relationship could be owing to the fact that these microorganisms are capable of both photosynthesis and nitrogen fixation, and that these biochemical processes could be required for the formation and progression of BBD. Seasonal representation of these organisms in BBD communities may reflect differences in their adaptation to water temperature, light intensity or other environmental conditions.
Mechanisms and signals associated with coral microbiome transitions to BBD remain unknown, although the detection of bacterial cell-to-cell QS signals (N-acyl homoserine lactones, AHLs and AI-2) within the BBD consortia (Zimmer et al., 2014) indicates that QS likely has a role in BBD communities. The ability to manipulate QS in associated microbiota is a common trait of marine organisms (Dobretsov et al., 2009). Therefore, it is, perhaps, not surprising that one of the most abundant cyanobacterial metabolites within BBD is a compound capable of manipulating QS in Vibrio spp.
By examining species co-occurrence patterns, we uncovered a negative correlation between Roseofilum and a number of bacteria that are present mostly in epibiomes. Negative co-occurrence patterns were also observed for Roseofilum and Vibrio species. Vibrios have previously been postulated to have a role in BBD (Barneah et al., 2007; Arotsker et al., 2009), although their contribution to the initiation and progression of the disease has yet to be characterized. Although vibrios were detected at moderately low levels in both healthy coral mucus and in the black band, vibrios are readily cultivable from both sample types and have QS activity through both the AHL-based autoinducer 1 system and the furanosyl borate diester-based autoinducer 2 system (Tait et al., 2010; Alagely et al., 2011a; Golberg et al., 2011, 2013; Zimmer et al., 2014). In addition to AHLs and AI-2, vibrios also produce hydroxyketone CAI-1 signals (Henke and Bassler, 2004; Ng et al., 2011; Wei et al., 2012), and with this study we demonstrate that coral vibrios also produce CAI-1-like activities. Furthermore, this CqsS/CAI-1 QS system appears vulnerable to manipulation by lyngbic acid, a cyanobacterial secondary metabolite.
Unlike previously characterized naturally occurring antagonists, which target AHL- or AI-2-based QS systems, activity of lyngbic acid required the presence of a functional CqsS, a receptor for the hydroxyketone CAI-1 signal. Synthetic agonists and antagonists of the CqsS-mediated signaling have been previously identified (Bolitho et al., 2011); however, lyngbic acid is the first characterized naturally occurring inhibitor of CqsS-mediated signaling. Even though lyngbic acid did not fully inhibit activity of the QS reporters (as was achieved by the some synthetic analogs of CAI-1), its activity at 16 nm is more potent than those reported for many of the synthetic antagonists (Bolitho et al., 2011). We note that deletion of the gene coding for the AHL receptor LuxN also reduced sensitivity of the strain to lyngbic acid at 2 μm, although not to the same extent as the deletion of the cqsS gene (Figure 5). It is possible that in addition to strongly inhibiting the CqsS-mediated cascades, lyngbic acid may also modestly affect the LuxN/AHL QS system, perhaps similarly to the ability of CAI-1 to antagonize AHL-based QS systems in other bacteria (Ganin et al., 2012). The consequences of manipulation of Vibrio QS with the cyanobacterial compound lyngbic acid are not yet known. In Vibrio spp., QS signals act as timers, with the CqsS/CAI-1 system active at low population densities (Henke and Bassler, 2004; Anetzberger et al., 2012). In concert with other QS signals, the CqsS/CAI-1 system regulates toxin and metalloprotease production, as well as type 3 secretion system (Henke and Bassler, 2004; Higgins et al., 2007). Therefore, functions of the cyanobacterial metabolite lyngbic acid may include manipulation of these behaviors to control interactions within BBD and its interactions with the host coral. This fundamental role for lyngbic acid as a QS inhibitor against vibrios may also explain its presence in so many different benthic filamentous marine cyanobacteria (Cardellina et al., 1978; Kwan et al., 2010; Soares et al., 2015).
Deep sequencing conducted in this study clearly establishes that Roseofilum reptotaenium, the cyanobacterial pathogen linked to BBD of corals, is a minor but ubiquitous member of healthy Caribbean coral microbiota. This observation has important implications for understanding how BBD initiates and spreads. Knowing that both Roseofilum and potentially pathogenic heterotrophs are in low abundance in healthy corals suggests that the disease consortium is held in check by currently unknown constraints exerted by either the coral polyp or the symbiotic algae, or the commensal Gammaproteobacteria (including Halomonas spp., Moritella spp.) and actinomycetes (Renibacterium spp.) characteristic of the healthy state. Release from these constraints is associated with the loss of the commensals (Halomonas spp., Moritella spp. and Renibacterium spp.) and an increase in the diversity and species richness of the microbiome. Even though R. reptotaenium is known to be the major member of the BBD consortium, BBD is the only example of a disease associated primarily with a cyanobacterium (Richardson et al., 2014). Given that cyanobacteria are not generally known for encoding genes involved in animal virulence, it is reasonable to hypothesize that the main function of R. reptotaenium in BBD is to act as an ‘engineer’ of the disease consortium: while not virulent on its own, through photosynthesis, nitrogen fixation and the biosynthesis of secondary metabolites, it creates an environment where opportunistic pathogens attack the coral host and degrade its tissues. To more robustly test this hypothesis, it will be important in future studies to characterize functions and chemicals that are involved in structuring of BBD.
References
Aeby GS, Santavy DL . (2006). Factors affecting susceptibility of the coral Montastreae faveolata to black-band disease. Mar Ecol Prog Ser 318: 103–110.
Alagely A, Krediet CJ, Ritchie KB, Teplitski M . (2011a). Signaling-mediated cross-talk modulates swarming and biofilm formation in a coral pathogen Serratia marcescens. ISME J 5: 1609–1620.
Alagely A, Rajamani S, Teplitski M . (2011b). Luminescent reporters and their applications for the characterization of signals and signal-mimics that alter LasR-mediated quorum sensing. Methods Mol Biol 692: 113–130.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic Local Alignment Search Tool. J Mol Biol 215: 403–410.
Anetzberger C, Reiger M, Fekete A, Schell U, Stambrau N, Plener L et al. (2012). Autoinducers act as biological timers in Vibrio harveyi. PLoS One 7: e48310.
Antonius A . (1973). New observations on coral destruction in reefs. Tenth Meeting of the Association of Island Marine Laboratories of the Caribbean Vol. 10. No. 3. University of Puerto Rico (Mayaguez).
Aronesty E . (2011). ea-utils: Command-line tools for processing biological sequencing data. Available from http://code.google.com/p/ea-utils.
Arotsker L, Siboni N, Ben-Dov E, Kramarsky-Winter E, Loya Y, Kushmaro A . (2009). Vibrio sp. as a potentially important member of the Black Band Disease (BBD) consortium in Favia sp. corals. FEMS Microb Ecol 70: 515–524.
Bader GD, Hogue CW . (2003). An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf 4: 2.
Barneah O, Ben-Dov E, Kramarsky-Winter E, Kushmaro A . (2007). Characterization of black band disease in Red Sea stony corals. Env Microbiol 9: 1995–2006.
Barott KL, Rodriguez-Brito B, Janouskovec J, Marhaver KL, Smith JE, Keeling P et al. (2011). Microbial diversity associated with four functional groups of benthic reef algae and the reef-building coral Montastraea annularis. Eniron Microbiol 13: 1192–1204.
Bassler BL, Wright M, Silverman MR . (1994). Multiple signalling systems controlling expression of luminescence in Vibrio harveyi: sequence and function of genes encoding a second sensory pathway. Mol Microbiol 13: 273–286.
Bayer T, Neave MJ, Alsheikh-Hussain A, Aranda M, Yum LK, Mincer T et al. (2013). The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl Environ Microb 79: 4759–4762.
Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M et al. (2010). Galaxy: a web-based genome analysis tool for experimentalists. Curr Prot Mol Biol Chapter 19: Unit 19.10.1–19.10.21.
Bolitho ME, Perez LJ, Koch MJ, Ng WL, Bassler BL, Semmelhack MF . (2011). Small molecule probes of the receptor binding site in the Vibrio cholerae CAI-1 quorum sensing circuit. Bioorg Med Chem 19: 6906–6918.
Bosch TC, McFall-Ngai MJ . (2011). Metaorganisms as the new frontier. Zoology (Jena) 114: 185–190.
Bourne DG, Garren M, Work TM, Rosenberg E, Smith GW, Harvell CD . (2009). Microbial disease and the coral holobiont. Trends Microbiol 17: 554–562.
Bruckner AW, Bruckner RJ, Williams EH . (1997). Spread of a black-band disease epizootic through the coral reef system in St Ann's Bay, Jamaica. B Mar Sci 61: 919–928.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7: 335–336.
Cardellina J, Dalietos D, Marner F, Mynderse J, Moore R . (1978). (−)-Trans-7(S-Methoxytetradec-4-enoic acid and related amides from the marine cyanophyte Lyngbya majuscula. Phytochemistry 17: 2091–2095.
Cardenas A, Rodriguez RL, Pizarro V, Cadavid LF, Arevalo-Ferro C . (2012). Shifts in bacterial communities of two Caribbean reef-building coral species affected by white plague disease. ISME J 6: 502–512.
Casamatta D, Stanic D, Gantar M, Richardson LL . (2012). Characterization of Roseofilum reptotaenium (Oscillatoriales, Cyanobacteria) gen. et sp nov isolated from Caribbean black band disease. Phycologia 51: 489–499.
Casey JM, Ainsworth TD, Choat JH, Connolly SR . (2014). Farming behaviour of reef fishes increases the prevalence of coral disease associated microbes and black band disease. Proc R Soc Ser B 281: 20141032.
Closek CJ, Sunagawa S, DeSalvo MK, Piceno YM, DeSantis TZ, Brodie EL et al. (2014). Coral transcriptome and bacterial community profiles reveal distinct Yellow Band Disease states in Orbicella faveolata. ISME J 8: 2411–2422.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl Environ Microb 72: 5069–5072.
Dixon P . (2003). VEGAN, a package of R functions for community ecology. J Veg Sci 14: 927–930.
Dobretsov S, Teplitski M, Paul V . (2009). Mini-review: quorum sensing in the marine environment and its relationship to biofouling. Biofouling 25: 413–427.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics (Oxford, England) 26: 2460–2461.
Edmunds PJ . (1991). Extent and effect of Black Band Disease on a Caribbean reef. Coral Reefs 10: 161–165.
Eren AM, Vineis JH, Morrison HG, Sogin ML . (2013). A filtering method to generate high quality short reads using Illumina paired-end technology. PLoS One 8: e66643.
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J et al. (2012). Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8: e1002606.
Frias-Lopez J, Zerkle AL, Bonheyo GT, Fouke BW . (2002). Partitioning of bacterial communities between seawater and healthy, black band diseased, and dead coral surfaces. Appl Environ Microbiol 68: 2214–2228.
Frias-Lopez J, Bonheyo GT, Jin Q, Fouke BW . (2003). Cyanobacteria associated with coral black band disease in Caribbean and Indo-Pacific Reefs. Appl Environ Microbiol 69: 2409–2413.
Ganin H, Danin-Poleg Y, Kashi Y, Meijler MM . (2012). Vibrio cholerae autoinducer CAI-1 interferes with Pseudomonas aeruginosa quorum sensing and inhibits its growth. ACS Chem Biol 7: 659–665.
Giardine B, Riemer C, Hardison R, Burhans R, Elnitski L, Shah P et al. (2005). Galaxy: a platform for interactive large-scale genome analysis. Gen Res 15: 1451–1455.
Glas MS, Sato Y, Ulstrup KE, Bourne DG . (2012). Biogeochemical conditions determine virulence of black band disease in corals. ISME J 6: 1526–1534.
Goecks J, Nekrutenko A, Taylor J, Team TG . (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Gen Biol 11: R86.
Golberg K, Eltzov E, Shnit-Orland M, Marks RS, Kushmaro A . (2011). Characterization of quorum sensing signals in coral-associated bacteria. Microb Ecol 61: 783–792.
Golberg K, Pavlov V, Marks RS, Kushmaro A . (2013). Coral-associated bacteria, quourum sensing disrupters, and the regulation of biofouling. Biofouling 29: 669–682.
Henke JM, Bassler BL . (2004). Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi. J Bacteriol 186: 6902–6914.
Higgins DA, Pomianek ME, Kraml CM, Taylor RK, Semmelhack MF, Bassler BL . (2007). The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature 450: 883–886.
Kuehl K, Jones R, Gibbs D, Richardson L . (2011). The roles of temperature and light in black band disease (BBD) progression on corals of the genus Diploria in Bermuda. J Invert Path 106: 366–370.
Kwan JC, Teplitski M, Gunasekera SP, Paul VJ, Luesch H . (2010). Isolation and biological evaluation of 8-epi-malyngamide C from the Floridian marine cyanobacterium Lyngbya majuscula. J Nat Prod 73: 463–466.
Littler MM, Littler DS . (1996). Black band disease in the South Pacific. Coral Reefs 15: 20–20.
Martin M . (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17: 10–12.
McKew BA, Dumbrell AJ, Daud SD, Hepburn L, Thorpe E, Mogensen L et al. (2012). Characterization of geographically distinct bacterial communities associated with coral mucus produced by Acropora spp. and Porites spp. Appl Environ Microb 78: 5229–5237.
McMurdie PJ, Holmes S . (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8: e61217.
Meyer JL, Paul VJ, Teplitski M . (2014). Community shifts in the surface microbiomes of the coral Porites astreoides with unusual lesions. PLoS One 9: e100316.
Miller AW, Blackwelder P, Al-Sayegh H, Richardson LL . (2011). Fine-structural analysis of black band disease-infected coral reveals boring cyanobacteria and novel bacteria. Dis Aquat Organ 93: 179–190.
Miller AW, Richardson LL . (2011). A meta-analysis of 16S rRNA gene clone libraries from the polymicrobial black band disease of corals. FEMS Microbiol Ecol 75: 231–241.
Miller MB, Bassler BL . (2001). Quorum sensing in bacteria. Ann Rev Microbiol 55: 165–199.
Mizrachi I . (2002) GenBank: the nucleotide sequence database. In: McEntyre J, Ostell J (eds). The NCBI Handbook [Internet], updated 22 August 2007 National Center for Biotechnology Information: Bethesda, MD, USA.
Montano S, Strona G, Seveso D, Galli P . (2013). Prevalence, host range, and spatial distribution of black band disease in the Maldivian Archipelago. Dis Aquat Organ 105: 65–74.
Morrow KM, Moss AG, Chadwick NE, Liles MR . (2012). Bacterial associates of two Caribbean coral species reveal species-specific distribution and geographic variability. Appl Environ Microbiol 78: 6438–6449.
Muller EM, van Woesik R . (2011). Black-band disease dynamics: prevalence, incidence, and acclimatization to light. J Exp Mar Biol Ecol 397: 52–57.
Ng WL, Bassler BL . (2009). Bacterial Quorum-Sensing network architectures. Annu Rev Genet 43: 197–222.
Ng WL, Perez LJ, Wei Y, Kraml C, Semmelhack MF, Bassler BL . (2011). Signal production and detection specificity in Vibrio CqsA/CqsS quorum-sensing systems. Mol Microbiol 79: 1407–1417.
Nübel U, Engelen B, Felske A, Snaidr J, Wieshuber A, Amann R et al. (1996). Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. Bacteriology 178: 5636–5643.
Page C, Willis B . (2006). Distribution, host range and large-scale spatial variability in black band disease prevalence on the Great Barrier Reef, Australia. Dis Aquat Organ 69: 41–51.
Rasoulouniriana D, Siboni N, Ben-Dov E, Kramarsky-Winter E, Loya Y, Kushmaro A . (2009). Pseudoscillatoria coralii gen. nov., sp. nov., a cyanobacterium associated with coral black band disease (BBD). Dis Aquat Organ 87: 91–96.
Richardson LL . (1998). Coral diseases: what is really known? Trends Ecol Evol 13: 438–443.
Richardson LL, Stanic D, May A, Brownell A, Gantar M, Campagna SR . (2014). Ecology and physiology of the pathogenic cyanobacterium Roseofilum reptotaenium. Life 4: 968–987.
Rideout JR, He Y, Navas-Molina JA, Walters WA, Ursell LK, Gibbons SM et al. (2014). Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2: e545.
Roder C, Arif C, Bayer T, Aranda M, Daniels C, Shibl A et al. (2014a). Bacterial profiling of White Plague Disease in a comparative coral species framework. ISME J 8: 31–39.
Roder C, Arif C, Daniels C, Weil E, Voolstra CR . (2014b). Bacterial profiling of White Plague Disease across corals and oceans indicates a conserved and distinct disease microbiome. Mol Ecol 23: 965–974.
Rosenberg E, Ben-Haim Y . (2002). Microbial diseases of corals and global warming. Environ Microbiol 4: 318–326.
Rypien KL, Ward JR, Azam F . (2010). Antagonistic interactions among coral-associated bacteria. Env Microbiol 12: 28–39.
Sato Y, Bourne DG, Willis BL . (2009). Dynamics of seasonal outbreaks of black band disease in an assemblage of Montipora species at Pelorus Island (Great Barrier Reef, Australia). Proc R Soc Ser B 276: 2795–2803.
Sato Y, Willis BL, Bourne DG . (2010). Successional changes in bacterial communities during the development of black band disease on the reef coral Montipora hispida. ISME J 4: 203–214.
Sato Y, Bourne DG, Willis BL . (2011). Effects of temperature and light on the progression of black band disease on the reef coral Montipora hispida. Coral Reefs 30: 753–761.
Sekar R, Mills DK, Remily ER, Voss JD, Richardson LL . (2006). Microbial communities in the surface mucopolysaccharide layer and the black band microbial mat of black band-diseased Siderastrea siderea. Appl Environ Microb 72: 5963–5973.
Sere MG, Tortosa P, Chabanet P, Turquet J, Quod JP, Schleyer MH . (2013). Bacterial communities associated with Porites White Patch Syndrome (PWPS) on three Western Indian Ocean (WIO) coral reefs. PLoS One 8: e83746.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Gen Res 13: 2498–2504.
Soares A, Engene N, Gunasekera S, Sneed J, Paul V . (2015). Carriebowlinol, an antimicrobial tetrahydroquinolinol from an assemblage of marine cyanobacteria containing a novel taxon. J Nat Prod 78: 534–538.
Sunagawa S, DeSantis TZ, Piceno YM, Brodie EL, DeSalvo MK, Voolstra CR et al. (2009). Bacterial diversity and white plague disease-associated community changes in the Caribbean coral Montastraea faveolata. ISME J 3: 512–521.
Sussman M, Bourne DG, Willis BL . (2006). A single cyanobacterial ribotype is associated with both red and black bands on diseased corals from Palau. Dis Aquat Organ 69: 111–118.
Tait K, Hutchison Z, Thompson FL, Munn CB . (2010). Quorum sensing signal production and inhibition by coral-associated vibrios. Environ Microbiol Rep 2: 145–150.
Teplitski M, Mathesius U, Rumbaugh KP . (2011). Perception and degradation of N-acyl homoserine lactone quorum sensing signals by mammalian and plant cells. Chem Rev 111: 100–116.
Vezzulli L, Pezzati E, Huete-Stauffer C, Pruzzo C, Cerrano C . (2013). 16SrDNA pyrosequencing of the Mediterranean Gorgonian Paramuricea clavata reveals a link among alterations in bacterial holobiont members, anthropogenic influence and disease outbreaks. PLoS One 8: e67745.
Wei Y, Ng WL, Cong J, Bassler BL . (2012). Ligand and antagonist driven regulation of the Vibrio cholerae quorum-sensing receptor CqsS. Mol Microbiol 83: 1095–1108.
Wickham H . (2009) ggplot2: Elegant Graphics for Data Analysis. Springer: : New York, NY, USA.
Zimmer BL, May AL, Bhedi CD, Dearth SP, Prevatte CW, Pratte Z et al. (2014). Quorum sensing signal production and microbial Interactions in a polymicrobial disease of corals and the coral surface mucopolysaccharide layer. PLoS One 9: e108541.
Zvuloni A, Artzy-Randrup Y, Stone L, Kramarsky-Winter E, Barkan R, Loya Y . (2009). Spatio-temporal transmission patterns of black-band disease in a coral community. PLoS One 4: e4993.
Acknowledgements
This research was supported by Protect Our Reefs Grant 2013-2, a George E Burch Fellowship in Theoretical Medicine and Affiliated Sciences at the Smithsonian Institution and the National Institutes of Health (R01 CA 172310). We are indebted to Dr B Bassler for sharing Vibrio harveyi QS reporters. Logistical support from the Center for Marine Studies, Honduras and funding from the Summit Foundation made possible sample collection off the Honduran coast. We thank E Bartels, C Thacker and C Walker for field assistance. This is a contribution 1007 of the Smithsonian Marine Station and contribution 979 from the Caribbean Coral Reef Ecosystems Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Meyer, J., Gunasekera, S., Scott, R. et al. Microbiome shifts and the inhibition of quorum sensing by Black Band Disease cyanobacteria. ISME J 10, 1204–1216 (2016). https://doi.org/10.1038/ismej.2015.184
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.184
This article is cited by
-
Coral and Seawater Metagenomes Reveal Key Microbial Functions to Coral Health and Ecosystem Functioning Shaped at Reef Scale
Microbial Ecology (2023)
-
Microbial dysbiosis reflects disease resistance in diverse coral species
Communications Biology (2021)
-
Identification of bacteria-derived urease in the coral gastric cavity
Science China Earth Sciences (2020)
-
Environmental flexibility in Oulastrea crispata in a highly urbanised environment: a microbial perspective
Coral Reefs (2020)
-
Marine probiotics: increasing coral resistance to bleaching through microbiome manipulation
The ISME Journal (2019)
