
Abstract
Mammalian cells remove misfolded proteins using various proteolytic systems, including the ubiquitin (Ub)-proteasome system (UPS), chaperone mediated autophagy (CMA) and macroautophagy. The majority of misfolded proteins are degraded by the UPS, in which Ub-conjugated substrates are deubiquitinated, unfolded and cleaved into small peptides when passing through the narrow chamber of the proteasome. The substrates that expose a specific degradation signal, the KFERQ sequence motif, can be delivered to and degraded in lysosomes via the CMA. Aggregation-prone substrates resistant to both the UPS and the CMA can be degraded by macroautophagy, in which cargoes are segregated into autophagosomes before degradation by lysosomal hydrolases. Although most misfolded and aggregated proteins in the human proteome can be degraded by cellular protein quality control, some native and mutant proteins prone to aggregation into β-sheet-enriched oligomers are resistant to all known proteolytic pathways and can thus grow into inclusion bodies or extracellular plaques. The accumulation of protease-resistant misfolded and aggregated proteins is a common mechanism underlying protein misfolding disorders, including neurodegenerative diseases such as Huntington’s disease (HD), Alzheimer’s disease (AD), Parkinson’s disease (PD), prion diseases and Amyotrophic Lateral Sclerosis (ALS). In this review, we provide an overview of the proteolytic pathways in neurons, with an emphasis on the UPS, CMA and macroautophagy, and discuss the role of protein quality control in the degradation of pathogenic proteins in neurodegenerative diseases. Additionally, we examine existing putative therapeutic strategies to efficiently remove cytotoxic proteins from degenerating neurons.
Similar content being viewed by others
Introduction
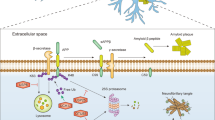
Misfolded proteins generated in various cellular compartments, including the cytoplasm, nucleus and endoplasmic reticulum (ER), are efficiently removed by quality control systems composed of the ubiquitin (Ub)-proteasome system (UPS), chaperone mediated autophagy (CMA) and macroautophagy (Figure 1).1 The first line of defense in degrading soluble misfolded proteins is the UPS (Figure 2), a selective proteolytic system in which substrates are tagged with Ub, unfolded into nascent polypeptide chains, and cleaved into short peptides while passing through the narrow chamber of the proteasome.1, 2, 3, 4 Specific misfolded proteins that expose the KFERQ degradation signal can be degraded by the CMA, a branch of the autophagy-lysosome system (hereafter autophagy), in which substrates are selectively recognized by the chaperone heat-shock cognate 70 (Hsc70) and directly delivered into lysosomes, leading to degradation by lysosomal hydrolases into amino acids (Figure 1).5, 6 Some misfolded proteins that escape the surveillance of the UPS and CMA or tend to form aggregates are directed to macroautophagy (Figure 1), a bulk degradation system in which substrates are segregated into autophagosomes which, in turn, are fused with lysosomes for degradation into amino acids (Figure 3).7, 8 Although almost all of the proteins encoded by the human genome can be efficiently removed from the cell when misfolded, a number of polypeptides generated from post-translational conjugation (for example, hyperphosphorylated tau in Alzheimer’s disease (AD)) or endoproteolytic cleavage (for example, amyloid β peptides) tend to be spontaneously misfolded and rapidly aggregated into oligomers enriched in β-sheet content.9, 10, 11, 12 Genetic mutations in specific proteins, such as huntingtin in Huntington’s disease (HD),13, 14 α-synuclein in Parkinson’s disease (PD),15, 16 prion protein (PrP) in prion diseases,17, 18, 19 and superoxide dismutase 1 (SOD1) and TAR DNA-binding protein 43 kDa (TDP-43) in Amyotrophic Lateral Sclerosis (ALS),20 may also perturb their folding, leading to the formation of similar β-sheet-enriched aggregates. The resulting oligomers are at least partially resistant to all known proteolytic pathways and can further grow into inclusion bodies or extracellular plaques that have highly ordered fibrillar structures with elevated β-sheet content.9 Cytotoxicity and neuronal death caused by misfolded oligomers and aggregates provide a molecular mechanism underlying the pathogenesis of many neurodegenerative diseases.21

The degradation of short-lived proteins by the UPS. In this selective proteolytic system, Ub is first activated by E1 and subsequently transferred to E2. In parallel, misfolded substrates of the UPS are recognized by molecular chaperones, such as CHIP, and associated with Ub ligases that promote the transfer of E2-conjugated Ub to specific Lys residues of substrates. Ubiquitinated substrates are deubiquitinated, unfolded, fed into the narrow chamber of the proteasome, and progressively cleaved into small peptides. Depending on the types of E3 ligases, Ub can be directly transferred from E2 to the substrate or via a two-step process that involves a transient binding of E3 to Ub. The repetition of this reaction results in the growth of a singly conjugated Ub to a chain of Ub with different topologies, depending on how Ub is conjugated to another Ub. Modified from Wang and Robbins.226

Autophagosome formation and lysosomal degradation. Autophagosome formation can be triggered when the mTOR complex is inhibited by various stressors, such as starvation. This induces the assembly of the ULK protein complex composed of ULK1, Atg13 and FIP200 at the isolation membrane, which, in turn, activates the formation of the Beclin-1/PI3KC3 complex composed of Beclin-1, UVRAG, Bif-1, Ambra1, Vps15 and Vps34. During the elongation of the isolation membrane, the Atg5-Atg12-Atg16L1 complex mediates the conjugation of PE to LC3-I, generating LC3-II that relocates from the cytosol to the autophagic membrane and is anchored on its surface. The resulting autophagic membrane structures—autophagosomes—are fused with lysosomes to form autolysosomes, wherein cargoes, including misfolded proteins, are degraded by lysosomal hydrolases.

The degradation of misfolded proteins by various cellular proteolytic pathways. Misfolded proteins are initially recognized by molecular chaperones that deliver the substrates to the UPS, CMA or macroautophagy depending on the nature of misfolding, size and solubility. In general, soluble and monomeric misfolded proteins are primarily degraded by the UPS and CMA. In CMA, substrates carrying the KFERQ motif are recognized and bound by Hsc70 in association with chaperones. The substrates are subsequently delivered to the LAMP2 complex on the lysosomal membrane, translocated to the lumen, and degraded into amino acids by lysosomal hydrolases. Some of these misfolded proteins tend to form aggregates and are thus directed to macroautophagy. Misfolded protein substrates of macroautophagy are recognized by molecular chaperones such as Hsc70, ubiquitinated by Ub ligases, and delivered to the autophagic adaptor p62, leading to the formation of p62 protein bodies. The targeted protein aggregates associated with p62 are subsequently delivered to autophagic membranes for lysosomal degradation, when p62 interacts with LC3 on the autophagic membrane.
Compared with proliferating cells, post-mitotic neurons are more sensitive to the accumulation of cytotoxic proteins because they cannot dilute toxic substances by means of cell division.22 Moreover, protein quality control is intrinsically challenging in neurons because of their unique cellular structure, characterized by the expansion of dendrites and axons in which protein aggregates need to be packaged into autophagic vacuoles and make a retrograde journey to the cell body, rich in lysosomes, for degradation.23, 24 Although young neurons can manage to clear cytotoxic proteins, this task becomes increasingly more difficult throughout the course of aging during which the components of the UPS, CMA and macroautophagy are downregulated in expression and activity.25, 26 In the affected neurons of many neurodegenerative diseases, such as AD, PD, HD, prion diseases and ALS, pathogenic protein aggregates can further downregulate the activities of proteolytic pathways.27, 28, 29, 30, 31, 32 One way to enhance degradation of pathogenic protein aggregates is to increase the activities of proteolytic pathways. Many small molecule compounds have been developed and successfully used to enhance the clearance of various pathogenic proteins.33, 34, 35, 36, 37, 38
The UPS in neurodegenerative diseases
The UPS is a proteolytic system in which the conjugation of Ub to substrates induces selective degradation by the proteasome (Figure 2).39 Protein degradation in the UPS is mediated by an enzymatic cascade composed of ~500–1000 proteins. In this ATP-consuming proteolytic system, Ub is first activated by forming a thioester bond between its C-terminal Gly76 residue and an active-site cysteine (Cys) of the Ub-activating enzyme E1. The activated Ub is transferred to the Ub-conjugating enzyme E2 via a thioester bond. It is the Ub ligase E3 that selectively recognizes and mediates ubiquitination of substrates, which involves the transfer of E2-conjugated Ub to lysine (Lys) residue(s) of the target substrate. The human genome is estimated to encode >500 E3 ligases, which can be classified into three groups depending on the types of ubiquitination domains, including the really interesting new gene (RING) finger, the homologous to E6-AP (HECT) domain and the U-box domain.40 An E3 Ub ligase can be a single polypeptide or a subunit of a protein complex, such as the SCF (Skp1-Cullin1-F-box) E3 complex. As Ub conjugation may occur at any of its seven Lys residues, a Ub chain can grow into many different topologies.41 The Lys48 linkage is the most widely used topology, which signals degradation by the proteasome, whereas the Lys63 linkage mediates non-proteolytic processes, such as Ub-dependent protein–protein interactions.42 The Lys11 linkage is typically used for cell-cycle regulation and cell division.43 Ub moieties on protein substrates can be removed by the deubiquitination enzyme to edit elongating chains or remove/recycle the targeted chains altogether from substrates.44, 45
Once ubiquitination generates a chain of four or more Ub at lysine 48, it can serve as a secondary degron that delivers the substrates to the 26S proteasome. This cylindrical machinery is composed of a proteolytic 20S core particle capped at both ends by a 19S regulatory particle.46, 47, 48 The 19S particle binds and unfolds the polyubiquitinated protein substrate and feeds the unfolded polypeptide chain into the chamber of the 20S particle, which is as narrow as 13 angstroms in diameter.47, 48 When feeding the substrates into the 20S particle, the 19S particle also deubiquitinates the polyubiquitinated substrates to recycle Ub. Passing through the 20S particle, the substrates are cleaved into small peptides by the β5, β2 and β1 subunits that have chymotrypsin-like, trypsin-like and caspase-like peptidase activities, respectively.47, 48
Substrates of the UPS include misfolded proteins, as well as a large number of short-lived proteins in the cytoplasm, nucleus, ER and other cellular compartments. The UPS-dependent degradation of misfolded proteins initiates when chaperones and Ub ligases recognize abnormalities in folding, such as hydrophobic residues exposed on the surface and improper disulfide bonds.49 Several E3s are known to mediate the ubiquitination of misfolded proteins. In the yeast Saccharomyces cerevisiae, the RING finger E3 ligase Ubr1, the recognition component of the N-end rule pathway, cooperates with chaperones to mediate the ubiquitination of misfolded cytosolic proteins for degradation by the proteasome.50, 51 The yeast Ub ligase San1 mediates the ubiquitination of misfolded proteins in the nucleus.52 With the help of heat-shock protein 70 (Hsp70), San1 also brings excessive cytosolic misfolded proteins to the nucleus for proteasomal degradation.51, 53 The yeast HECT Ub ligase Hul5 was recently found to mediate the ubiquitination of misfolded proteins generated by heat shock.54 In mammals, the U-box-containing E3 ligase CHIP is known to interact with Hsp70 and promote the delivery of misfolded cytosolic proteins to cellular degradation machinery.55 Little is known about the mammalian Ub ligases involved in quality control of misfolded proteins in neurons.
The pathogenesis of many neurodegenerative diseases, including AD, PD, ALS, HD and prion diseases, is associated with and, moreover, at least partly contributed by the downregulation of the UPS.56, 57 One major risk factor underlying reduced UPS activities in degenerating brains is aging. Extensive studies have shown that proteasomal activities can gradually decrease with aging, which results in a reduced capacity to degrade misfolded proteins, contributing to the formation of pathological protein aggregates.27, 28, 29, 31 Another risk factor is the presence of aggregated proteins that inhibit the activities of UPS components, including the proteasome. For example, aggregated β-sheet-rich PrP blocks the opening of the 20S proteasome particle, leading to reduced proteasomal activity.58 Ubiquitinated and aggregated tau in AD can block the gate of the 19S catalytic particle by binding to its recognition site, leading to a traffic jam and impaired proteasomal degradation.30, 32 In addition, recent studies have shown that aggregates of many other pathogenic proteins in neurodegenerative disorders can directly inhibit proteasome activity.59, 60, 61, 62
The autophagy-lysosome system in neurodegenerative diseases
Autophagy is a process by which cytoplasmic constituents are degraded by the lysosome. Protein quality control via autophagy is particularly important for the timely removal of aggregated forms of pathogenic proteins in neurodegenerative diseases, including tau in AD, α-synuclein in PD and polyQ-Htt in HD.63, 64 Autophagy can be divided into microautophagy, CMA and macroautophagy, depending on the mechanism by which cellular cargoes are delivered to the lysosome (Figure 1).65 Among the three arms of autophagy, the targeted clearance of misfolded proteins is mainly mediated by CMA and macroautophagy. CMA is a selective proteolytic system in which specific misfolded proteins carrying the KFERQ motif are delivered to and degraded in lysosomes. This pentapeptide motif, found in ~30% of cytosolic proteins, is normally buried by protein folding, but it can be exposed on the surface by misfolding or partial unfolding. It is recognized by the chaperone Hsc70 associated with cochaperones.6 The substrates are subsequently delivered to the CMA adaptor (lysosomal membrane-associated protein 2A (LAMP-2A) on the lysosomal membrane, unfolded, translocated into the lysosomal lumen and degraded into amino acids. In degenerating neurons, CMA can be constitutively activated to compensate for impaired macroautophagy.66
In macroautophagy, a portion of cytoplasmic constituents, such as misfolded proteins and organelles, are segregated by double-membrane structures called autophagosomes and subsequently digested by lysosomal hydrolases (Figure 1). The delivery of misfolded proteins to autophagosomes involves specific adaptors, including the p62/SQSTM-1/sequestosome.67 The autophagic adaptor p62 has a UBA (Ub association) domain that interacts with polyubiquitin chains of misfolded proteins and a PB1 domain that mediates self-aggregation to form condensed cargo-p62 complexes.68, 69, 70 Cargo-loaded p62 and its aggregated complexes are delivered to autophagic vacuoles through the specific interaction of p62 with light chain 3 II (LC3-II), an active form of LC3, on the surface of autophagic double membrane structures.71 By inducing aggregation and eventually delivery to autophagic vacuoles, p62 reduces the toxicity of a free form or oligomeric species of misfolded proteins destined for macroautophagy.72 Mutations in the p62 gene have been implicated in the pathogenesis of Paget disease of bone as well as familial and sporadic ALS.73 In addition to p62, other autophagic adaptors, such as NBR1, NDP52, optineurin (OPTN), histone deacetylase 6 and NIX26, mediate the delivery of various types of cellular cargoes to autophagic membranes through similar mechanisms.74, 75 Once misfolded proteins are loaded to phagophores, the autophagic membrane structures are fused with each other to grow into autophagosomes, which are fused in turn with lysosomes, generating autolysosomes in which cargoes are degraded by lysosomal hydrolases. Autophagosome formation involves a large number of proteins and their post-translational modifications, such as the ATG7-mediated conjugation of ATG5 (autophagy-related protein 5) to ATG12, leading to cleavage and lipidation of LC3-I to form LC3-II (Figure 3).7, 76, 77 Upon conversion, cytosolic LC3-II is translocated to autophagic membranes and acts as an anchor to receive cargoes through interaction with autophagic adaptors.
Although misfolded proteins can be immediately and directly delivered to autophagosomes, excess misfolded or damaged proteins and their aggregates that accumulate beyond cellular capacity are temporarily stored in the aggresome, a cytoplasmic inclusion in the microtubule organizing center near the nucleus.9 During this process, called aggrephagy, the histone deacetylase 6, in association with molecular chaperones, binds freely floating ubiquitinated aggregates and delivers them via microtubules to a location that minimizes their toxicity until they are finally degraded by the UPS or macroautophagy.78, 79, 80, 81 The major components of aggresomes include ubiquitinated proteins as well as specific regulatory proteins involved in the formation and degradation of proteins aggregates, such as p62, ALFY (autophagy-linked FYVE protein) and NBR1 (neighbor of BRCA1 gene).
The functions and survival of neurons heavily depend on the efficient removal of misfolded proteins by autophagy because they cannot dilute cytotoxic proteins by cell division. In addition, autophagy is an intrinsically challenging process in neurons because of their unique cellular structure characterized by the expansion of dendrites and axons. For example, misfolded proteins that have been generated in axons and nerve terminals are packaged on site into autophagosomes and make a long retrograde journey to the cell body, wherein lysosomes are enriched in the perinuclear microtubule-organizing center.22 Before reaching the cell body, autophagosomes in the process of retrograde transportation often fuse with late endosomes generated in neurites, resulting in the formation of amphisomes.23, 24 This is a time-consuming, difficult and complicated process whose overall efficiency can be adversely affected by many factors, such as aging and genetic mutations. Extensive studies have shown that many components of CMA and macroautophagy are downregulated at the levels of transcription, translation and post-translation as neurons age.25, 26 These age-sensitive regulators include the substrate recognizer/carrier Hsc7082, 83 and the Hsc70-acceptor LAMP-2A in CMA84 as well as Beclin-1 in macroautophagy.85, 86 Reduced autophagic activity appears to be pharmaceutically manageable, as the restoration of CMA by maintaining LAMP-2A levels in aging mouse livers has been shown to promote liver health and increase the ability of hepatocytes to degrade damaged proteins.84 In addition to reduced autophagic activity in aged neurons, the activities of autophagic components can be adversely affected by interaction with protein aggregates,87, 88, 89 which can be excessively generated by age-dependent impairment of the UPS. For example, tau in frontotemporal lobar dementia with Ub-positive inclusions and α-synuclein in PD bind LAMP-2A with an unusually high affinity, leading to a traffic jam during cargo translocation across the lysosomal membrane.89 Yet another risk factor underlying dysregulation of autophagy in aged neurons is a genetic mutation in a regulator of autophagy, such as p62, whose mutations are implicated in the pathogenesis of familial and sporadic ALS32, characterized by p62-positive inclusions in affected neurons.90
Protein quality control in AD: Aβ and tau
AD is the most common form of progressive dementia, characterized by cognitive impairment, memory loss and behavioral abnormalities. This protein misfolding disorder is caused by the misfolding and aggregation of amyloid β peptides and tau, which give rise to amyloid plaques and neurofibrillary tangles, respectively.91 Aβ is a 42-residue product resulting from two sequential cleavages of the amyloid precursor protein (APP), a transmembrane protein with no clearly defined function. The first cleavage produces a C-terminal fragment, and the fragment is then cleaved by the γ-secretase complex composed of presenilin-1, APH-1, PEN-2 and nicastrin92 to generate Aβ, which tends to be misfolded to form aggregates.10, 11 Mutations of various genes, including APP, can upregulate the production of Aβ, contributing to the pathogenesis of AD.10, 11 By contrast, APP and Aβ can be downregulated by the UPS at various steps of processing, from the ER lumen to the plasma membrane.93 The first UPS degradation occurs after a nascent APP polypeptide is cotranslationally translocated into the ER lumen, during which its signal peptide is cleaved off. Following translocation, a successfully folded APP mature protein enters the Golgi secretory pathway. However, terminally misfolded APP is degraded via ER-associated degradation in which substrates are unfolded, ubiquitinated, retrotranslocated across the ER membrane and degraded by the proteasome. The targeting by ER-associated degradation involves the E3 Ub ligases HRD194 and Fbxo2.95 Proteasomal degradation can also occur when APP arrives at the Golgi apparatus, where APP is ubiquitinated though a K63 linkage by unknown E3 ligases stimulated by ubiquilin-1, leading to the retention of APP without proteasomal degradation.96 Even after being presented at the plasma membrane, APP can be internalized to endosomes and enter the endosome-Golgi pathway, where APP can be cleaved to generate Aβ.97 The resulting intracellular Aβ is prone to misfolding and is targeted by UPS-dependent protein quality control, which includes the E3 ligase CHIP that mediates the ubiquitination of misfolded proteins for proteasomal degradation.93 In contrast to APPs, however, Ub-conjugated Aβ in affected neurons is not properly degraded through the proteasome.98
Recent studies have implicated autophagy in the turnover of Aβ. In an AD mouse model overexpressing Aβ, haploinsufficiency of Beclin-1 reduced autophagy and exacerbated AD pathology, as evidenced by Aβ deposition and neurodegeneration, which was rescued by lentiviral administration of Beclin-1.99 Conditional mutant mice lacking ATG7 in the central nervous system showed degeneration of pyramidal neurons in the hippocampus and Purkinje cells in the cerebellum.100 Genetic inactivation of other autophagic components in neurons, such as ATG5 or ATG17/FIP200, resulted in similar neuronal degeneration.25, 101 While the turnover of Aβ involves autophagy, autophagy itself is impaired in the brains of AD patients. For example, affected neurons in AD brains are enriched in autophagosomes and other types of autophagic vacuoles that together act as a major intracellular reservoir of cytotoxic peptides.102 The excessive accumulation of immature autophagic vacuoles in senile neurons is associated with increased synthesis of autophagic core components, retrograde transportation of autophagosomes and impaired fusion with lysosomes, contributing to the accumulation of pathogenic Aβ.103, 104
Another hallmark of AD is neurofibrillary tangles composed primarily of phosphorylated tau.105 Although neurofibrillary tangles were initially thought to be one of the major causes of AD pathogenesis,106 recent studies indicate that a monomeric form of tau with pathological modifications and its soluble oligomers may be more cytotoxic.12 The tau protein can lose its function through various proteolytic events, including cleavage by endoproteolytic enzymes such as caspases,107 calpain,108 aminopeptidases109 and thrombin.110 However, these cleavages are unlikely to contribute to the clearance of neurofibrillary tangles because the resulting cleavage products with various modifications may aid the development of AD. The first line of defense against tau accumulation is the E3 ligase CHIP, which mediates the ubiquitination of tau (primarily in its phosphorylated form), in collaboration with Hsp70 and Hsp90 (Figure 4).111 An in vitro study showed that the E2 enzyme Ube2w can also mediate E3-independent ubiquitination of tau.112 However, ubiquitinated tau is not a good substrate of the proteasome and thus accumulates as detergent-resistant aggregates, leading to the formation of neurofibrillary tangles in AD. In the process of targeting tau to the proteasome, CHIP also appears to be deposited to neurofibrillary tangles with its substrate and other ubiquitinated proteins.98, 111 It has been shown that UPS-dependent clearance of tau is facilitated by overexpressing the molecular chaperone Hsp70, which binds misfolded proteins.111 As UPS-dependent degradation of tau is not efficient, autophagy has a close relationship with AD pathogenesis with respect to the formation of amyloid plaques and tau aggregates.113 For example, autophagic inhibition by 3-methylamphetamine or cloroquine was shown to slow tau clearance, leading to tau aggregation.114 By contrast, rapamycin, an inducer of autophagy, inhibited the accumulation of tau aggregates and neurotocixity using a mouse tau model.37 Pharmaceutical inhibition of phospholipase D1, which regulates autophagosome maturation downstream of Vps34, resulted in neuronal accumulation of tau and p62 aggregates.115 A subpopulation of caspase-generated tau fragments has been shown to be delivered to autophagic vacuoles.116 Defective autophagic flux promotes the formation of tau oligomers and insoluble aggregates. A phosphorylated form of tau shows reduced binding to microtubules and bundling as well as an increased tendency to be found as motile particles.117

The degradation of tau proteins. Tau can be targeted by both the UPS and macroautophagy, depending on the nature of post-translational modifications that influence folding and solubility. In general, soluble monomeric tau proteins are recognized by molecular chaperones and Ub ligases, such as CHIP, leading to the formation of ubiquitinated tau proteins. It remains unclear as to what extent ubiquitinated tau proteins are actually degraded by the proteasome. Alternatively, the same substrates can be directly delivered to the 20S proteasome without ubiquitination. Some tau proteins prone to rapid aggregation, such as hyperphosphorylated species, can be delivered to p62 and, subsequently, autophagosomes for lysosomal degradation. Modified from Chesser et al.227
Protein quality system in PD: α-synuclein
PD is the most common neurodegenerative movement disorder. It is characterized by decreased motor ability and the loss of dopaminergic neurons in the substantia nigra pars compacta. The major pathogenic agent of PD is a mutant form of α-synuclein, a presynaptic nerve terminal protein.118 The activity of mutant α-synuclein as an autosomal dominant cause for PD is associated with point mutations (for example, A53T, A30P and E46K) that render α-synuclein prone to misfolding and aggregation.119, 120, 121 The accumulation of aggregated mutant α-synuclein leads to the formation of intracellular inclusions called Lewy bodies (LBs), which serve as the major hallmarks of both sporadic and familial PD. In addition to mutant α-synuclein, LBs contain more than 90 proteins, including PD markers (DJ-1, LRRK2 (leucine-rich repeat kinase 2), Parkin and PINK-1 (PTEN-induced putative kinase 1)) and mitochondria-related proteins, as well as components of the UPS and autophagy, particularly those involved in aggresome formation.122, 123, 124 Consistent with the finding that many of the proteins accumulated in LBs are involved in protein quality control, major causative mutations in familial PD are linked to genes in the UPS or autophagic pathways, including α-synuclein, PINK-1, the Ub ligase Parkin, UCH-L1 (Ub carboxy terminal hydrolase L1), DJ-1 (PARK7) and LRRK2/PRAK8.122 In the pathogenesis of PD, monomeric and non-fibrillar mutant α-synuclein molecules may be more cytotoxic than fibrillar aggregates, and LBs found in the brains of PD patients may be a consequence of cytoprotective responses.122, 123, 124
Wild-type α-synuclein has been shown to be ubiquitinated and degraded by the proteasome using in vitro assays89, 125, 126 and cultured neuronal cells under proteasomal inhibition.127, 128, 129 However, other studies have suggested that ubiquitination is not needed for proteasomal degradation of α-synuclein (Figure 5).130, 131 Proteasomal degradation of α-synuclein has been shown to be facilitated by its phosphorylation at Ser129.132 Several regulatory proteins of the UPS were implicated in the turnover of soluble α-synuclein in the cytosol, including Ub ligases CHIP,133 SIAH,134, 135 MDM2136 and HRD1.137 A subpopulation of α-synuclein associated with membranes in the endosome-lysosome pathway has been shown to be targeted by the Ub ligase Nedd4.138 In addition to Ub ligases, rare mutations in the deubiquitinating enzyme UCH-L1 have been associated with familial, early onset PD.139 PD-linked mutants of UCH-L1 contain only partial deubiquitinating activities, contributing to the accumulation of α-synuclein in presynaptic terminals.140 The role of UCH-L1 in PD pathogenesis is in part attributed to its activity as an E3 ligase, whereby it mediates K63-linked ubiquitination in its dimer form.141 The overall importance of the UPS in the turnover of α-synuclein is further supported by the finding that conditional knockout mice lacking Psmc1, a proteasomal subunit, in nigral or forebrain neurons resulted in the formation of intraneuronal LB-like inclusions positive for Ub and α-synuclein associated with neurodegeneration.142 While soluble α-synuclein is degraded by the proteasome, its filamentous form can interact directly with the 20S core of the proteasome and decrease its proteolytic activity.61 Consistently, proteasome misregulation has been observed in the substantia nigra of PD patients.143

The degradation of α-synuclein by cellular protein quality control. Wild-type and mutant α-synuclein can be targeted by the ubiquitination-dependent UPS (A) and possibly in a manner independent from Ub (B) as well. Monomeric α-synuclein can also be targeted by the CMA (C). By contrast, macroautophagy can degrade monomeric and oligomeric α-synuclein as well as its aggregates (D). Intracellular α-synuclein can also be cleaved by endopeptidases, such as calpains (E) and neurosin (F). Extracellular α-synuclein can be cleaved by neurosin (G) and metalloproteinases (H). The resulting proteolytic cleavage products are thought to contribute to the cytotoxicity of α-synuclein. Modified from Xilouri et al.228
Recent studies have shown that α-synuclein can be degraded by CMA through a specific CMA recognition motif.89, 144 However, the A30P and A53T PD-linked mutants have unusually high affinity for the CMA adaptor LAMP-2A and are not efficiently delivered to the lysosomal lumen, resulting in a traffic jam in CMA.89, 145 This, in turn, can trigger compensating macroautophagy.146 The hydrolysis of CMA-targeted α-synuclein in the lysosomal lumen involves cathepsin D, a primary lysosomal protease.147, 148 Although α-synuclein in a monomeric or soluble oligomeric form can be targeted by both the UPS and the CMA, its aggregates are directed to the lysosome via macroautophagy. The role of macroautophagy in α-synuclein degradation was suggested by the finding that α-synuclein is accumulated in the lysosome of cultured neuronal cells under macroautophagic inhibition, whereas the lysosomal targeting of PD-linked mutant α-synuclein was attenuated under the same conditions.33, 149 Pharmacological activation of macroautophagy using rapamycin, a mammalian target of rapamycin (mTOR) inhibitor, facilitated the degradation of both wild-type and mutant α-synuclein.138, 150 The clearance of α-synuclein by macroautophagy was further shown in transgenic mice virally overexpressing Beclin-1, an autophagic regulator.150
Protein quality control in HD: mutant huntingtin proteins
HD is an autosomal dominant neurodegenerative disorder that affects ~5–10 individuals per 100 000.151 Affected individuals suffer from progressive motor and cognitive declines associated with loss of self and spatial awareness, depression, dementia and increased anxiety. This progressive neurodegenerative disease is caused by the aggregation of mutant huntingtin (mHTT) proteins. The wild-type huntingtin protein (HTT) contains a stretch of the glutamine residue, called polyQ tract, which is encoded by a repeat of the codon CAG within exon 1 of the HTT gene.152, 153 The length of the CAG repeat varies between individuals and generations, ranging on average between 16 and 20 repeats.154 In affected individuals, the CAG repeat expands to >35 in number, giving rise to the elongated polyQ tract of mHTT proteins that are prone to aggregation and toxic to neurons.14, 155 PolyQ inclusions are abundant in highly ordered amyloid fibers with enriched β-sheets and low detergent solubility.156 PolyQ inclusions may be a consequence of a protective mechanism to sequester small oligomeric forms of mHTT, which are highly cytotoxic to neurons.157 Extracellular polyQ aggregates can be internalized by cells to initiate a new round of polyQ aggregation, suggesting that mHTT may act as an infectious agent through a mechanism observed in prion diseases.158
Despite the importance of mHTT in the pathogenesis of HD, surprisingly little is known about the mechanism by which cytotoxic mHTT is removed from the cell. This is perhaps because mHTT is a poor substrate for all known proteolytic pathways, including UPS, CMA, and macroautophagy. Moreover, extensive studies have shown that mHTT acts as an inhibitor of proteolytic machineries, often in the process of its turnover.159 For example, mHTT inclusions in the brains of HD patients and HD mice are enriched in the components of the UPS, such as Ub and ubiquitinated HTT, because mHTT species can be initially tagged with Ub but are poor substrates for the proteasome.160 It has been suggested that the accumulation of mHTT inclusions is not a consequence of direct proteasomal inhibition but rather result from the gross failure of protein quality control systems in association with the sequestration of molecular chaperones.161
Wild-type HTT can be degraded by CMA,162 during which Hsc70 recognizes two KFERQ-like motifs, KDRVN at residues 99–103 and NEIKV at residues 248–252.159 Like HTT, mHTT can also be recognized by Hsc70 for CMA degradation.159 However, the polyQ expansion of mHTT delays the delivery of mHTT across the lysosomal membrane because mHTT has a higher affinity for Hsc70 and LAMP-2A.159 Failure to promptly deliver the initially targeted mHTT to the lysosome results in a traffic jam in CMA-dependent autophagic degradation, leading to a secondary side effect in proteostasis. Failure to degrade mHTT results in the accumulation of perinuclear cytoplasmic aggregates and intranuclear inclusions in the neurons of patients with HD.162
Core components of macroautophagy, such as LC3, are typically upregulated in various HD mouse models and in neuronal and non-neuronal cells in patients with HD.163, 164 The apparent upregulation of macroautophagy is associated with the excessive formation of cargo-free autophagic vacuoles, possibly because the delivery of cargoes to autophagic vacuoles is impaired.163 As the autophagic flux is reduced, components of macroautophagy, such as p62, LC3-II, mTOR and Beclin-1, were found to be deposited in the striatum of HD transgenic mice.165 The sequestration of autophagic regulators in mHTT inclusions, such as mTOR, contributes to the increased synthesis of autophagic core components.85, 166 Thus, HD disease progression is exacerbated by reduced activities of macroautophagy associated with HTT inhibition of macroautophagy in an age-dependent manner.
Protein quality control in prion diseases: scrapie prion protein
Prion diseases, also known as transmissible spongiform encephalopathies, are infectious neurodegenerative disorders in humans and animals that affect the brain and nervous system, leading to spongiform vacuolation and severe neuronal loss.167 Prion diseases in animals include nature scrapie in sheep and goat,168 bovine spongiform encephalopathy (also known as mad cow disease) in cattle,169 chronic wasting disease in elk and deer,170 and feline spongiform encephalopathy in domestic cats.171 In humans, these fatal protein misfolding disorders include kuru172, Creutzfeldt–Jakob disease173, Gerstmann–Sträussler–Scheinker syndrome174, fatal familial insomnia175 and new variant CJD (a human equivalent to bovine spongiform encephalopathy/mad cow disease).167
The transmissible agent common to these transmissible diseases is scrapie prion protein (PrPSc), an abnormally misfolded isoform of the host-encoded cellular prion protein (PrPC).18 PrPC is a glycosylphosphatidyl inositol-linked glycoprotein enriched in α-helical structure. This cell surface protein with no clearly defined function is initially translated as a 253-residue polypeptide and enters the ER wherein its signal peptide is cleaved off, generating a 208-residue mature protein. The mature PrPC polypeptide undergoes folding and is conjugated with sugar moieties during transportation via the Golgi-secretory pathway. In this process, a soluble form of misfolded PrPC is normally degraded by various protein quality control systems, including Ub-dependent ER-associated degradation. Compared with PrPC, however, PrPSc is enriched in β-sheets and tends to form aggregates that are at least partially resistant to all known cellular protein quality control systems.17, 18, 19 Moreover, PrPSc can interact with PrPC and facilitate the conversion of PrPC into PrPSc, which, in turn, can convert more PrPC into PrPSc, resulting in the accumulation of misfolded and aggregated PrPSc in the brain.176, 177, 178 Through this seeding-nucleation process, a small quantity of invading PrPSc is enough to trigger the autocatalytic conversion of host PrPC into PrPSc.179, 180 The transmissible nature of PrPSc has been demonstrated by the finding that the inoculation of small quantities of PrPSc into animals led to characteristics of prion diseases.177, 178
Compared with the clinical importance of PrPSc, surprisingly little is known of its turnover. In principle, as the conversion of PrPC into PrPSc requires significant refolding and conformational changes in folding, this process may involve chaperones and Ub ligases of UPS-dependent protein quality control. Indeed, recent studies in S. cerevisiae suggest that Hsp70, Hsp40 and Hsp26 may loosen prion fibrils, whereas Hsp104 fully disassembles the fibrils into shorter fragments.181 In mammalian cells, the chaperones GroEL and Hsp104 were shown to facilitate the conversion of PrPC into PrPSc in the presence of a small amount of PrPSc that served as a seed.182 In addition, Hsc70, a recognition component of CMA, was shown to bind to PrPC.183 Despite the implication of chaperones in the turnover of PrPC, it appears that PrPSc is not a good substrate of the UPS. Moreover, recent studies have shown that PrPSc binds to the 20S proteasome without further processing and thus blocks substrate entry into the proteolytic chamber, leading to proteasomal failure.62, 184 PrPSc may also bind to the external surface of the 20S particle and induce an allosteric stabilization of the closed state of the 20S proteasome.58, 185 Consistent with these findings, prion diseases are associated with impaired activities of the UPS.185 As a consequence of proteasomal inhibition, cellular Ub conjugates are excessively accumulated in mouse brain infected with ME7 scrapie train.185
Prion diseases are associated with misregulation of autophagy as evidenced by the formation of giant autophagic vacuoles in experimental scrapie in hamsters.186 These autophagic vacuoles often grow in size and number as neurons age, eventually occupying the entire volume of the affected neurites.187 The formation of giant autophagic vacuoles is caused by the reduced flux of autophagy in combination with endosomal/lysosomal dysfunction, which may contribute to the pathogenesis of prion diseases.187 Although a study showed that recombinant PrPC mutants (V203I, E211Q and Q212P) overexpressed in neuroblastoma cells were converted to PrPSc-like aggregates and delivered to aggresomes,188 there is no evidence that PrPSc is efficiently processed by autophagic pathways. Instead, recent studies indicate that prion proteins adversely affect autophagy, as exemplified by the finding that the overexpression of a PrPC-like protein, Doppel (Dpl), in neurons resulted in the progressive death of Purkinje cells in prion-lacking Ngsk mice.189 As further described in the following sections, one way to facilitate the clearance of PrPSc is to use small molecules that stimulate autophagy.35, 190, 191
Protein quality control in ALS: SOD and TDP-43
ALS is a progressive paralytic disease characterized by selective degeneration and death of motor neurons associated with the accumulation of misfolded proteins and insoluble inclusions.20 Although indistinguishable in clinical symptoms, this protein misfolding disorder can be divided into sporadic ALS, which accounts for ~82% of all ALS cases, and familial ALS.20 Mutations in ALS may occur in genes encoding key components of protein quality control. This group of mutant ALS proteins includes dynein and dynactin, both involved in the retrograde transport of autophagosomes from axons to the cell body,192, 193 the autophagic adaptor p62,73 and the UBA-containing proteins Ubqln2 and Optineurin.194 Another group of ALS mutations generates proteins with abnormal folding, leading to aggregation and the formation of insoluble inclusions.20 This latter group includes SOD1, TDP-43, and FUS/TLS (Fused in Sarcoma/Translocated in Sarcoma).20, 195 Approximately 20% of familial ALS cases are caused by over 140 different point mutations of SOD1, a soluble cytosolic enzyme that dismutates superoxide radicals to H2O2.196 SOD1 mutants are mostly dominant and causative to the death of affected motor neurons because they tend to be misfolded and form protease-resistant aggregates.195 Another ASL-relevant gene is TDP-43, in which mutations account for ~5% of sporadic ALS and 3% of familial ALS cases.20 This hnRNP family member can bind to RNA in a single-stranded and sequence-specific manner, which is required for many RNA processes.197 One unique aspect of TDP-43 is the property of its C-terminal tail to be prone to misfolding and aggregation.197, 198 Like other pathogenic mutant proteins in neurodegenerative diseases, misfolded SOD1 and TDP-43 mutants are initially targeted for degradation by the components of the UPS, such as chaperones and Ub ligases.20 Owing to their tendency to aggregate, however, the targeted mutants escape during the delivery process to the proteasome, some of which are redirected to autophagy. ALS mutants resistant to the UPS and autophagy are aggregated together to form intracellular inclusions containing Ub and Ub ligases found in familial ALS mutant mice199, 200 and post-mortem spinal cord of sporadic ALS patients.201, 202, 203 It was reported that the insoluble inclusions typically become visible in the brain stem and spinal cord at the onset of ALS symptoms and progressively accumulate throughout late stages.204 Although large inclusions are clinical hallmarks of ALS symptoms, they are unlikely to be toxic to neurons. They may, however, be a neuroprotective phenomenon, as it was suggested that monomeric and oligomeric misfolded ALS proteins are the actual toxic substance in motor neurons.195
Autophagy is often misregulated in the spinal cord of sporadic ALS patients, as evidenced by the excessive formation of autophagosomes.205 The autophagic misregulation can be partially explained by findings stating that inclusions observed in ALS patients can impair protein quality controls by sequestering various components ranging from proteasomal subunits and Ub ligases, such as Dorfin, to molecular chaperones HSP70 and HSP40 and the motor protein dynein involved in cargo delivery to the aggresome.5, 206, 207 Monomeric or oligomeric ALS proteins can also directly inhibit both proteasomal activity197, 198, 208, 209 and autophagic flux.210, 211, 212 Moreover, it has been shown that reduced proteasomal activity can promote the accumulation of ALS protein aggregates.213 Thus, one mechanism underlying the pathogenesis of ALS is a vicious cycle between misfolded proteins and proteolytic pathways, which accelerates the excessive accumulation of insoluble inclusions, leading to the death of affected motor neurons.
Targeting autophagy for therapy of neurodegenerative diseases
Substantial benefits of therapy could be achieved with agents that promote the degradation of pathogenic proteins underlying neurodegenerative diseases. Many small molecules that induce autophagy have been developed and shown to be effective in removing pathogenic proteins. The therapeutic activities of the autophagic inducer rapamycin, an inhibitor of mTOR, have been demonstrated using transgenic mouse models of neurodegenerative diseases, such as AD mice expressing mutant APP,36, 38 AD mice expressing tau,37 HD mice expressing mHTT,214 PD mice expressing mutant α-synuclein33 and prion disease mice expressing PrPSc.35 The overall results indicate that rapamycin promotes the clearance of these pathogenic protein aggregates, improves cognition and behavior and ameliorates neuropathology and neurodegeneration in the brains of these transgenic mouse models. Similar therapeutic benefits were obtained using analogs of rapamycin, such as CCI-779, which was shown to reduce mHTT aggregates, leading to improved motor behaviors in HD transgenic mice.215 In contrast, rapamycin worsened autophagic functions and neuron degeneration in a SOD1(G93A) transgenic mouse model of ALS212 and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxin models of PD,216 suggesting that autophagic induction may exert adverse effects on certain neurodegenerative conditions.
Various autophagic inducers were exploited to enhance the clearance of pathogenic protein aggregates in neurodegenerative diseases by targeting the ULK1 kinase AMP-activated protein kinase (AMPK) or cAMP–inositol 1,4,5-trisphosphate.13 An mTOR-independent macroautophagy inducer, Rilmenidine, was shown to improve motor ability and the clearance of mHTT fragment in transgenic HD mice.217 The mood stabilizer lithium, known to inhibit inositol monophosphatase and the phosphoinositol cycle, promoted the degradation of various protein aggregates including PrPSc of prion disease,191 mHTT of HD, α-synuclein of PD218 and SOD1 G93A of ALS.219, 220 Trehalose is a natural disaccharide product with pharmacological chaperone activity that exerts a protective role against various environmental stresses.221 This mTOR-independent autophagy activator was shown to enhance the clearance of mHTT in cultured cells, reduce the toxicity of mHTT and improve motor ability and lifespan in transgenic HD mice.221, 222 Trehalose promoted the clearance of A30P and A53T α-synuclein mutants in cultured PD model cells.221 The natural flavone finsetin and related compounds that activate autophagy through both target of rapamycin complex 1 (TORC1) and AMPK activities showed protective effects in neurodegenerative models.223 Protein phosphatase 2A agonists that inhibit tau hyperphosphorylation and activate autophagy through TORC1 and AMPK are under clinical trials for AD.224 Not surprisingly, a synergistic effect was obtained when rapamycin and Trehalose were combined to remove pathogenic protein aggregates of HD and PD.221 The combination of rapamycin and the IMPase inhibitor lithium was also shown to reduce the toxicity of mHTT.34 These results suggest that the combination therapy based on an mTOR inhibitor and an mTOR-independent activator may need to be further exploited for therapeutic application, although off-target effects are expected to increase. Collectively, these studies demonstrated that autophagic inducers have potential as therapeutic agents for selected neurodegenerative diseases. The overall effects of these reagents on a broad range of biological processes in neurons and non-neuronal cells require further investigation.
Concluding remarks
It is estimated that there will be two billion people over the age of 60 by 2050. One common biochemical mechanism underlying most neurodegenerative disorders is the failure of protein quality control to degrade or remove misfolded proteins in the brains of aged persons. The disease-causing misfolded proteins are generated over the course of aging by post-translational modifications (for example, endoproteolytic cleaves and phosphorylation) of native proteins (for example, amyloid β and tau in AD) or genetic mutations of otherwise non-pathogenic proteins (for example, HTT in HD, α-synuclein in PD, PrPC in prion disease and SOD1 and TDP-43 in ALS). These pathogenic agents tend to aggregate into oligomers with enriched β-sheet content, which can further grow into fibrillar inclusion bodies or extracellular plaques, serving as clinical hallmarks of many neurodegenerative diseases. β-Sheet-enriched aggregates can impair—either directly or indirectly—the UPS as well as CMA and macroautophagy by interacting with various cellular molecules, including key components of proteolytic pathways. This results in the reduced ability of protein quality control, which further accelerates the accumulation of cytotoxic aggregates. This exacerbating cycle between misfolded proteins and protein quality control is particularly toxic to aged neurons as the ability of these post-mitotic cells to cope with such difficulties is naturally reduced over the course of aging. As a consequence of these unfortunate events, neurodegenerative diseases are typically associated with global failures of all proteolytic pathways.
A significant portion of cellular proteins is misfolded during translation/folding or while functioning as folded proteins, either spontaneously or under cellular stresses. Most abnormally folded cellular proteins in the human proteome can be efficiently removed through the cooperative work of the UPS, CMA and macroautophagy. In contrast, the aforementioned pathogenic proteins are commonly resistant to those proteolytic pathways, perhaps because their β-sheet-enriched folds are difficult for molecular chaperones to loosen up. These substrates, without being fully unfolded, cannot be properly fed into the proteasomal cylinder, may be stuck within the narrow cylinder of the proteasome, or may not readily dissociate from the components (for example, LAMP-2) of CMA while being delivered across the lysosomal membrane. One strategy to enhance the clearance of pathogenic proteins is to enhance the activities or levels of molecular chaperones engaged in the UPS, as demonstrated by a study in which the overexpression of the molecular chaperone Hsp70 accelerated the proteasomal degradation of tau.111 Another strategy is to activate the molecular chaperones (for example, Hsc70), carriers (for example, histone deacetylase 6) and/or adaptors (for example, LAMP-2) of CMA, as a few studies have shown that the augmentation of CMA enhanced the removal of pathogenic misfolded proteins.8, 159, 225 One common limitation of the UPS and CMA is that substrates should be at least partially or completely unfolded into nascent polypeptides before they are fed into the proteasome or lysosome. By contrast, the degradation by macroautophagy does not involve an ATP-dependent unfolding step, making this lysosomal proteolysis an ideal quality control system for aggregation-prone misfolded proteins. In addition, although autophagic flux is often reduced in affected neurons in most neurodegenerative diseases, the functions of core autophagic machinery appear to remain largely intact, as several studies have shown that the alteration of autophagic regulators such as mTOR fully restored the autophagic flux. As such, many small molecule compounds were developed to induce macroautophagy and demonstrated to enhance the clearance of cytotoxic protein aggregates. As the mTOR pathway is emerging as a promising drug target, known mTOR-dependent autophagic inducers were successfully used to enhance the clearance of various pathogenic protein aggregates, improve cognition and behavior, and ameliorate neurodegeneration in the brains of various transgenic mouse models. Other regulators of autophagy, such as the ULK1 kinase AMPK, are also being actively exploited as potential drug targets, with synergistic effects between rapamycin and an mTOR-independent autophagic inducer. Although it is increasingly clear that autophagy inducers have therapeutic potential to remove protein aggregates, it should be noted that most of these studies use transgenic mice overexpressing pathogenic proteins that have already formed high levels of insoluble inclusions. The activities of these compounds on a broad range of biological processes, including off-target effects, should be further investigated under more physiologically relevant conditions.
References
Ciechanover A . Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Bioorg Med Chem 2013; 21: 3400–3410.
Sriram SM, Kim BY, Kwon YT . The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Biol 2011; 12: 735–747.
Tasaki T, Sriram SM, Park KS, Kwon YT . The N-end rule pathway. Annu Rev Biochem 2012; 81: 261–289.
Kim ST, Tasaki T, Zakrzewska A, Yoo YD, Sung KS, Kim BY et al. The N-end rule proteolytic system in autophagy. Autophagy 2013; 9: 1100–1103.
Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J et al. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet 2010; 19: 3219–3232.
Kiffin R, Christian C, Knecht E, Cuervo AM . Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 2004; 15: 4829–4840.
Hariharan N, Zhai P, Sadoshima J . Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal 2011; 14: 2179–2190.
Koga H, Cuervo AM . Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol Dis 2011; 43: 29–37.
Kopito RR . Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 2000; 10: 524–530.
Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 19: 353–356.
Huang Y, Mucke L . Alzheimer mechanisms and therapeutic strategies. Cell 2012; 148: 1204–1222.
Ward SM, Himmelstein DS, Lancia JK, Binder LI . Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans 2012; 40: 667–671.
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4: 295–305.
Tsoi H, Lau TC, Tsang SY, Lau KF, Chan HY . CAG expansion induces nucleolar stress in polyglutamine diseases. Proc Natl Acad Sci USA 2012; 109: 13428–13433.
Martin I, Dawson VL, Dawson TM . Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet 2011; 12: 301–325.
Uversky VN . Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J Neurochem 2007; 103: 17–37.
Griffith JS . Self-replication and scrapie. Nature 1967; 215: 1043–1044.
Prusiner SB . Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136–144.
Prusiner SB . Prions. Proc Natl Acad Sci USA 1998; 95: 13363–13383.
Andersen PM, Al-Chalabi A . Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nature Rev Neurol 2011; 7: 603–615.
Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG . Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 2005; 280: 17294–17300.
Lee S, Sato Y, Nixon RA . Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci 2011; 31: 7817–7830.
Hollenbeck PJ . Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol 1993; 121: 305–315.
Larsen KE, Sulzer D . Autophagy in neurons: a review. Histol Histopathol 2002; 17: 897–908.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441: 885–889.
Mizushima N . The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22: 132–139.
Keller JN, Huang FF, Markesbery WR . Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 2000; 98: 149–156.
Jung KM, Astarita G, Zhu C, Wallace M, Mackie K, Piomelli D . A key role for diacylglycerol lipase-alpha in metabotropic glutamate receptor-dependent endocannabinoid mobilization. Mol Pharmacol 2007; 72: 612–621.
Tydlacka S, Wang CE, Wang X, Li S, Li XJ . Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci 2008; 28: 13285–13295.
Dantuma NP, Lindsten K . Stressing the ubiquitin-proteasome system. Cardiovascular Research 2010; 85: 263–271.
Löw K, Aebischer P . Use of viral vectors to create animal models for Parkinson's disease. Neurobiol Dis 2012; 48: 189–201.
Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT . The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol 2012; 181: 1426–1435.
Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC . Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003; 278: 25009–25013.
Sarkar S, Krishna G, Imarisio S, Saiki S, O'Kane CJ, Rubinsztein DC . A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum Mol Genet 2008; 17: 170–178.
Heiseke A, Aguib Y, Riemer C, Baier M, Schätzl HM . Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem 2009; 109: 25–34.
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S . Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem 2010; 285: 13107–13120.
Rodriguez-Navarro JA, Cuervo AM . Autophagy and lipids: tightening the knot. Semin Immunopathol 2010; 32: 343–353.
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010; 5: e9979.
Hershko A, Ciechanover A . The ubiquitin system. Annu Rev Biochem 1998; 67: 425–479.
Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C . CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 2006; 440: 551–555.
Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 2003; 21: 921–926.
Hadian K, Griesbach RA, Dornauer S, Wanger TM, Nagel D, Metlitzky M et al. NF-kappaB essential modulator (NEMO) interaction with linear and lys-63 ubiquitin chains contributes to NF-kappaB activation. J Biol Chem 2011; 286: 26107–26117.
Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, Bustos D et al. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell 2010; 39: 477–484.
Sowa ME, Bennett EJ, Gygi SP, Harper JW . Defining the human deubiquitinating enzyme interaction landscape. Cell 2009; 138: 389–403.
Komander D, Rape M . The ubiquitin code. Annu Rev Biochem 2012; 81: 203–229.
Clague MJ, Urbé S . Ubiquitin: same molecule, different degradation pathways. Cell 2010; 143: 682–685.
Hendil KB, Khan S, Tanaka K . Simultaneous binding of PA28 and PA700 activators to 20 S proteasomes. Biochem J 1998; 332: 749–754.
Tanahashi N, Murakami Y, Minami Y, Shimbara N, Hendil KB, Tanaka K . Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J Biol Chem 2000; 275: 14336–14345.
Ravid T, Hochstrasser M . Diversity of degradation signals in the ubiquitin–proteasome system. Nat Rev Mol Cell Biol 2008; 9: 679–690.
Eisele F, Wolf DH . Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett 2008; 582: 4143–4146.
Heck JW, Cheung SK, Hampton RY . Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci USA 2010; 107: 1106–1111.
Fredrickson EK, Rosenbaum JC, Locke MN, Milac TI, Gardner RG . Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Mol Biol Cell 2011; 22: 2384–2395.
Prasad R, Kawaguchi S, Ng DT . A nucleus-based quality control mechanism for cytosolic proteins. Mol Biol Cell 2010; 21: 2117–2127.
Fang NN, Ng AH, Measday V, Mayor T . Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat Cell Biol 2011; 13: 1344–1352.
Connell CM, Shaw BA, Holmes SB, Foster NL . Caregivers' attitudes toward their family members' participation in Alzheimer disease research: implications for recruitment and retention. Alzheimer Dis Assoc Disord 2001; 15: 137–145.
Hegde AN, Upadhya SC . Role of ubiquitin-proteasome mediated proteolysis in nervous system disease. Biochim Biophys Acta 2011; 1809: 128–140.
Dennissen FJ, Kholod N, van Leeuwen FW . The ubiquitin proteasome system in neurodegenerative diseases: culprit, accomplice or victim? Prog Neurobiol 2012; 96: 190–207.
Andre R, Tabrizi SJ . Misfolded PrP and a novel mechanism of proteasome inhibition. Prion 2012; 6: 32–36.
Gregori L, Fuchs C, Figueiredo-Pereira ME, Van Nostrand WE, Goldgaber D . Amyloid -protein inhibits ubiquitin-dependent protein degradation in vitro. J Biol Chem 1995; 270: 19702–19708.
Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B . Aggregated and monomeric α -synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J Biol Chem 2003; 278: 11753–11759.
Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB et al. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem 2004; 279: 12924–12934.
Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell 2007; 26: 175–188.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimerrelated PS1 mutations. Cell 2010; 141: 1146–1158.
Nixon RA, Yang DS, Lee JH . Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 2008; 4: 590–599.
Kaushik S, Cuervo AM . Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012; 22: 407–417.
Kaushik S, Massey AC, Mizushima N, Cuervo AM . Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 2008; 19: 2179–2192.
Deretic V . Autophagy in infection. Curr Opin Cell Biol 2010; 22: 252–262.
Ichimura Y, Kominami E, Tanaka K, Komatsu M . Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 2008; 4: 1063–1066.
Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283: 22847–22857.
Ichimura Y, Komatsu M . Selective degradation of p62 by autophagy. Semin Immunopathol 2010; 32: 431–436.
Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 2010; 38: 265–279.
Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol 2010; 191: 537–552.
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2011; 68: 1440–1446.
Deretic V . A master conductor for aggregate clearance by autophagy. Dev Cell 2010; 18: 694–696.
Johnson CW, Melia TJ, Yamamoto A . Modulating macroautophagy: a neuronal perspective. Future Med Chem 2012; 4: 1715–1731.
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y . In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15: 1101–1111.
Perry CN, Kyoi S, Hariharan N, Takagi H, Sadoshima J, Gottlieb RA . Novel methods for measuring cardiac autophagy in vivo. Methods Enzymol 2009; 453: 325–342.
Liu XD, Ko S, Xu Y, Fattah EA, Xiang Q, Jagannath C et al. Transient aggregation of ubiquitinated proteins is a cytosolic unfolded protein response to inflammation and endoplasmic reticulum stress. J Biol Chem 2012; 287: 19687–19698.
Wong ES, Tan JM, Soong WE, Hussein K, Nukina N, Dawson VL et al. Autophagy mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet 2008; 17: 2570–2582.
Kirilyuk A, Shimoji M, Catania J, Sahu G, Pattabiraman N, Giordano A et al. An intrinsically disordered region of the acetyltransferase p300 with similarity to prion-like domains plays a role in aggregation. PLoS One 2012; 7: e48243.
Johnston JA, Ward CL, Kopito RR . Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998; 143: 1883–1898.
Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ et al. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009; 323: 124–127.
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 2010; 67: 1464–1472.
Zhang C, Cuervo AM . Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 2008; 14: 959–965.
Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T et al. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 2006; 281: 14474–14485.
David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C . Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biology 2010; 8: e1000450.
Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 2009; 18: 4153–4170.
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 2013; 16: 394–406.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D . Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004; 305: 1292–1295.
Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N . Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44: 279–289.
Jiang T, Yu JT, Tian Y, Tan L . Epidemiology and etiology of Alzheimer’s disease: from genetic to non-genetic factors. Curr Alzheimer Res 2013; 10: 852–867.
De Strooper B . Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 2003; 38: 9–12.
Kumar P, Ambasta RK, Veereshwarayya V, Rosen KM, Kosik KS, Band H et al. CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence Abeta metabolism. Hum Mol Genet 2007; 16: 848–864.
Kaneko M, Koike H, Saito R, Kitamura Y, Okuma Y, Nomura Y . Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J Neurosci 2010; 30: 3924–3932.
Atkin G, Hunt J, Minakawa E, Sharkey L, Tipper N, Tennant W et al. F-box only protein 2 (Fbxo2) regulates amyloid precursor levels and processing. J Biol Chem 2014; 289: 7038–7048.
El Ayadi A, Stieren ES, Barral JM, Boehning D . Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63-linked polyubiquitination of lysine 688. Proc Natl Acad Sci USA 2012; 109: 13416–13421.
Thinakaran G, Koo EH . Amyloid precursor protein trafficking, processing, and function. J Biol Chem 2008; 283: 29615–29619.
Perry G, Friedman R, Shaw G, Chau V . Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci USA 1987; 84: 3033–3036.
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA et al. The autophagy related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008; 118: 2190–2199.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441: 880–884.
Liang CC, Wang C, Peng X, Gan B, Guan JL . Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem 2010; 285: 3499–3509.
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A et al. Extensive involvement of autophagy in Alzheimer disease: an immunoelectron microscopy study. J Neuropathol Exp Neurol 2005; 64: 113–122.
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci 2008; 28: 6926–6937.
Nixon RA, Yang DS . Autophagy failure in Alzheimer’s diseasedlocating the primary defect. Neurobiol Dis 2011; 43: 38–45.
Haass C, Selkoe DJ . Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nature Rev Mol Cell Biol 2007; 8: 101–112.
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J 2012; 26: 1946–1959.
Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci USA 2003; 100: 10032–10037.
Canu N, Dus L, Barbato C, Ciotti MT, Brancolini C, Rinaldi AM et al. Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J Neurosci 1998; 18: 7061–7074.
Karsten SL, Sang TK, Gehman LT, Chatterjee S, Liu J, Lawless GM et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron 2006; 51: 549–560.
Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z et al. Inducible expression of tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem 2006; 281: 1205–1214.
Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet 2004; 13: 703–714.
Scaglione KM, Basrur V, Ashraf NS, Konen JR, Elenitoba-Johnson KS, Todi SV et al. The ubiquitin-conjugating enzyme (E2) Ube2w ubiquitinates the N terminus of substrates. J Biol Chem 2013; 288: 18784–18788.
Lee MJ, Lee JH, Rubinsztein DC . Tau degradation: the ubiquitin- proteasome system versus the autophagy-lysosome system. Prog Neurobiol 2013; 105: 49–59.
Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C et al. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci 2008; 27: 1119–1130.
Dall'Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB et al. The phospholipase D1 pathway modulates macroautophagy. Nat Commun 2010; 1: 142.
Metcalfe MJ, Huang Q, Figueiredo-Pereira ME . Coordination between proteasome impairment and caspase activation leading to TAU pathology: neuroprotection by cAMP. Cell Death Dis 2012; 3: e326.
Rodríguez-Martín T, Cuchillo-Ibáñez I, Noble W, Nyenya F, Anderton BH, Hanger DP . Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging 2013; 34: 2146–2157.
Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995; 14: 467–475.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997; 276: 2045–2047.
Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet 1998; 18: 106–108.
Singleton AB, Farrer M, Johnson J, Singleton A, Haque S, Kachergus J et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003; 302: 841.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M . α-Synuclein in Lewy bodies. Nature 1997; 388: 839–840.
Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM et al. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 1998; 152: 879–884.
Seidel K, Schöls L, Nuber S, Petrasch-Parwez E, Gierga K, Wszolek Z et al. First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann Neurol 2010; 67: 684–689.
Liu CW, Corboy MJ, DeMartino GN, Thomas PJ . Endoproteolytic activity of the proteasome. Science 2003; 299: 408–411.
Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338: 949–953.
Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM . Degradation of alpha-synuclein by proteasome. J Biol Chem 1999; 274: 33855–33858.
Imai Y, Soda M, Takahashi R . Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem 2000; 275: 35661–35664.
McLean PJ, Kawamata H, Hyman BT . Alpha-synucleinenhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 2001; 104: 901–912.
Tofaris GK, Layfield R, Spillantini MG . Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 2001; 509: 22–26.
Nakajima T, Takauchi S, Ohara K, Kokai M, Nishii R, Maeda S et al. Alpha-synuclein-positive structures induced in leupeptin-infused rats. Brain Res 2005; 1040: 73–80.
Machiya Y, Hara S, Arawaka S, Fukushima S, Sato H, Sakamoto M et al. Phosphorylated alpha-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin independent manner. J Biol Chem 2010; 285: 40732–40744.
Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ . The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 2005; 280: 23727–23734.
Liani E, Eyal A, Avraham E, Shemer R, Szargel R, Berg D et al. Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson's disease. Proc Natl Acad Sci USA 2004; 101: 5500–5505.
Lee JT, Wheeler TC, Li L, Chin LS . Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum Mol Genet 2008; 17: 906–917.
Nair VD, McNaught KS, González-Maeso J, Sealfon SC, Olanow CW . p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. J Biol Chem 2006; 281: 39550–39560.
Mei J, Niu C . Alterations of Hrd1 expression in various encephalic regional neurons in 6-OHDA model of Parkinson's disease. Neurosci Lett 2010; 474: 63–68.
Tofaris GK, Kim HT, Hourez R, Jung JW, Kim KP, Goldberg AL . Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal–lysosomal pathway. Proc Natl Acad Sci USA 2011; 108: 17004–17009.
Das C, Hoang QQ, Kreinbring CA, Luchansky SJ, Meray RK, Ray SS et al. Structural basis for conformational plasticity of the Parkinson's disease-associated ubiquitin hydrolase UCH-L1. Proc Natl Acad Sci USA 2006; 103: 4675–4680.
Cartier AE, Ubhi K, Spencer B, Vazquez-Roque RA, Kosberg KA, Fourgeaud L et al. Differential effects of UCHL1 modulation on alpha-synuclein in PD-like models of alpha-synucleinopathy. PLoS One 2012; 7: e34713.
Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT Jr. . The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell 2002; 111: 209–218.
Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and lewy-like inclusions resembling human pale bodies. J Neurosci 2008; 28: 8189–8198.
McNaught KS, Jenner P . Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett 2001; 297: 191–194.
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L . Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 2008; 283: 23542–23556.
Malkus KA, Ischiropoulos H . Regional deficiencies in chaperone-mediated autophagy underlie alpha-synuclein aggregation and neurodegeneration. Neurobiol Dis 2012; 46: 732–744.
Shen YF, Tang Y, Zhang XJ, Huang KX, Le WD . Adaptive changes in autophagy after UPS impairment in Parkinson’s disease. Acta Pharmacol Sin 2013; 34: 667–673.
Sevlever D, Jiang P, Yen SH . Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008; 47: 9678–9687.
Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain 2009; 2: 5.
Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM et al. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci 2001; 21: 8053–8061.
Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci 2009; 29: 13578–13588.
Munoz-Sanjuan I, Bates GP . The importance of integrating basic and clinical research toward the development of new therapies for Huntington disease. J Clin Invest 2011; 121: 476–483.
Walker FO . Huntington's disease. Lancet 2007; 369: 218–228.
Arrasate M, Finkbeiner S . Protein aggregates in Huntington’s disease. Exp Neurol 2012; 238: 1–11.
Warby SC, Visscher H, Collins JA, Doty CN, Carter C, Butland SL et al. HTT haplotypes contribute to differences in Huntington disease prevalence between Europe and East Asia. Eur J Hum Genet 2011; 19: 561–566.
Williams AJ, Paulson HL . Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 2008; 31: 521–528.
Soto C . Unfolding the role of protein misfolding in neurodegenerative diseases. Nature Rev Neurosci 2003; 4: 49–60.
Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol 2011; 7: 925–934.
Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R . Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat Struct Mol Biol 2011; 18: 328–336.
Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M et al. The role of chaperone–mediated autophagy in huntingtin degradation. PLoS ONE 2012; 7: e46834.
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997; 277: 1990–1993.
Hipp MS, Patel CN, Bersuker K, Riley BE, Kaiser SE, Shaler TA et al. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J Cell Biol 2012; 196: 573–587.
Jeong H, Then F, Melia TJ Jr., Mazzulli JR, Cui L, Savas JN et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009; 137: 60–72.
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 2010; 13: 567–576.
Wong E, Cuervo AM . Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010; 13: 805–811.
Lee H, Noh JY, Oh Y, Kim Y, Chang JW, Chung CW et al. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet 2012; 21: 101–114.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36: 585–595.
Kovacs GG, Budka H . Prion diseases: from protein to cell pathology. Am J Pathol 2008; 172: 555–565.
Ma J, Wang F . Prion disease and the ‘protein-only hypothesis’. Essays Biochem 2014; 56: 181–191.
Wells GA, Scott AC, Johnson CT, Gunning RF, Hancock RD, Jeffrey M et al. A novel progressive spongiform encephalopathy in cattle. Vet Rec 1987; 121: 419–420.
Williams ES, Young S . Spongiform encephalopathy of Rocky Mountain elk. J Wildl Dis 1982; 18: 465–471.
Wyatt JM, Pearson GR, Smerdon TN, Gruffydd-Jones TJ, Wells GA, Wilesmith JW . Naturally occurring scrapie-like spongiform encephalopathy in five domestic cats. Vet Rec 1991; 129: 233–236.
Gajdusek DC, Gibbs CJ, Alpers M . Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature 1966; 209: 794–796.
Gibbs CJ Jr., Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM et al. Creutzfeldt–Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 1968; 161: 388–389.
Masters CL, Gajdusek DC, Gibbs CJ Jr. . Creutzfeldt–Jakob disease virus isolations from the Gerstmann–Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain 1981; 104: 559–588.
Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A et al. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 1986; 315: 997–1003.
Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ et al. Synthetic mammalian prions. Science 2004; 305: 673–676.
Castilla J, Saá P, Hetz C, Soto C . In vitro generation of infectious scrapie prions. Cell 2005; 121: 195–206.
Wang F, Wang X, Yuan CG, Ma J . Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327: 1132–1135.
Saborio GP, Permanne B, Soto C . Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411: 810–813.
Soto C . Transmissible proteins: expanding the prion heresy. Cell 2012; 149: 968–977.
Shorter J, Lindquist S . Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 2004; 304: 1793–1797.
DebBurman SK, Raymond GJ, Caughey B, Lindquist S . Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc Natl Acad Sci USA 1997; 94: 13938–13943.
Wilkins S, Choglay AA, Chapple JP, van der Spuy J, Rhie A, Birkett CR et al. The binding of the molecular chaperone Hsc70 to the prion protein PrP is modulated by pH and copper. Int J Biochem Cell Biol 2010; 42: 1226–1232.
Förster A, Masters EI, Whitby FG, Robinson H, Hill CP . The 1.9 Å structure of a proteasome-11S activator complex and implications for proteasome-PAN/PA700 interactions. Mol Cell 2005; 18: 589–599.
Deriziotis P, André R, Smith DM, Goold R, Kinghorn KJ, Kristiansen M et al. Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J 2011; 30: 3065–3077.
Boellaard JW, Kao M, Schlote W, Diringer H . Neuronal autophagy in experimental scrapie. Acta Neuropathol 1991; 82: 225–228.
Sikorska B, Liberski PP, Brown P . Neuronal autophagy and aggresomes constitute a consistent part of neurodegeneration in experimental scrapie. Folia Neuropathol 2007; 45: 170–178.
Mishra RS, Bose S, Gu Y, Li R, Singh N . Aggresome formation by mutant prion proteins: the unfolding role of proteasomes in familial prion disorders. J Alzheimers Dis 2003; 5: 15–23.
Heitz S, Grant NJ, Bailly Y . Doppel induces autophagic stress in prion protein-deficient Purkinje cells. Autophagy 2009; 5: 422–424.
Heiseke A, Aguib Y, Schatzl HM . Autophagy, prion infection and their mutual interactions. Curr Issues Mol Biol 2010; 12: 87–97.
Aguib Y, Heiseke A, Gilch S, Riemer C, Baier M, Schatzl HM et al. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009; 5: 361–369.
Moughamian AJ, Holzbaur EL . Dynactin is required for transport initiation from the distal axon. Neuron 2012; 74: 331–343.
Ikenaka K, Kawai K, Katsuno M, Huang Z, Jiang YM, Iguchi Y et al. dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS One 2013; 8: e54511.
Fecto F, Siddique T . UBQLN2/P62 cellular recycling pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Muscle Nerve 2012; 45: 157–162.
Guo Y, Li C, Wu D, Wu S, Yang C, Liu Y et al. Ultrastructural diversity of inclusions and aggregations in the lumbar spinal cord of SOD1-G93A transgenic mice. Brain Res 2010; 1353: 234–244.
Beleza-Meireles A, Al-Chalabi A . Genetic studies of amyotrophic lateral sclerosis: controversies and perspectives. Amyotroph Lateral Scler 2009; 10: 1–14.
Buratti E, Baralle FE . Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci 2008; 13: 867–878.
Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD . TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 2009; 284: 20329–20339.
Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997; 18: 327–338.
Bendotti C, Atzori C, Piva R, Tortarolo M, Strong MJ, Debiasi S et al. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J Neuropathol Exp Neurol 2004; 63: 113–119.
Leigh PN, Whitwell H, Garofalo O, Buller J, Swash M, Martin JE et al. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 1991; 114: 775–788.
Mendonca DM, Chimelli L, Martinez AM . Expression of ubiquitin and proteasome in motorneurons and astrocytes of spinal cords from patients with amyotrophic lateral sclerosis. Neurosci Lett 2006; 404: 315–319.
Sasaki S . Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 2010; 69: 346–355.
Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD . Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 2001; 8: 933–941.
Morimoto N, Nagai M, Ohta Y, Miyazaki K, Kurata T, Morimoto M et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res 2007; 1167: 112–117.
Di Noto L, Whitson LJ, Cao X, Hart PJ, Levine RL . Proteasomal degradation of mutant superoxide dismutases linked to amyotrophic lateral sclerosis. J Biol Chem 2005; 280: 39907–39913.
Hoffman EK, Wilcox HM, Scott RW, Siman R . Proteasome inhibition enhances the stability of mouse Cu/Zn superoxide dismutase with mutations linked to familial amyotrophic lateral sclerosis. J Neurol Sci 1996; 139: 15–20.
Hyun DH, Lee M, Halliwell B, Jenner P . Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins. J Neurochem 2003; 86: 363–373.
Puttaparthi K, Wojcik C, Rajendran B, DeMartino GN, Elliott JL . Aggregate formation in the spinal cord of mutant SOD1 transgenic mice is reversible and mediated by proteasomes 1. J Neurochem 2003; 87: 851–860.
Carra S, Crippa V, Rusmini P, Boncoraglio A, Minoia M, Giorgetti E et al. Alteration of protein folding and degradation in motor neuron diseases: implications and protective functions of small heat shock proteins. Prog Neurobiol 2012; 97: 83–100.
Carra S, Rusmini P, Crippa V, Giorgetti E, Boncoraglio A, Naujock N et al. Different anti-aggregation and pro-degradative functions of the members of the mammalian sHSP family in neurological disorders. Philos Trans R Soc Lond B Biol Sci 2013; 368: 20110409.
Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X et al. Rapamycin treatment augments motor neuron degeneration in SOD1G93A mouse model of amyotrophic lateral sclerosis. Autophagy 2011; 7: 412–425.
Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E et al. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet 2010; 19: 3440–3456.
Sarkar S, Rubinsztein DC . Huntington's disease: degradation of mutant huntingtin by autophagy. FEBS J 2008; 275: 4263–4270.
Ravikumar B, Duden R, Rubinsztein DC . Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002; 11: 1107–1117.
Domanskyi A, Geissler C, Vinnikov IA, Alter H, Schober A, Vogt MA et al. Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson’s disease models. FASEB J 2011; 25: 2898–2910.
Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadig O et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet 2010; 19: 2144–2153.
Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170: 1101–1111.
Feng HL, Leng Y, Ma CH, Zhang J, Ren M, Chuang DM . Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008; 155: 567–572.
Pizzasegola C, Caron I, Daleno C, Ronchi A, Minoia C, Carrì MT et al. Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph Lateral Scler 2009; 10: 221–228.
Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC . Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant Huntingtin and alpha-synuclein. J Biol Chem 2007; 282: 5641–5652.
Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 2004; 10: 148–154.
Maher P, Dargusch R, Ehren JL, Okada S, Sharma K, Schubert D et al. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS One 2011; 6: e21226.
Magnaudeix A, Wilson CM, Page G, Bauvy C, Codogno P, Lévêque P et al. PP2A blockade inhibits autophagy and causes intraneuronal accumulation of ubiquitinated proteins. Neurobiol Aging 2013; 34: 770–790.
Jia H, Kast RJ, Steffan JS, Thomas EA . Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: implications for the ubiquitin-proteasomal and autophagy systems. Hum Mol Genet 2012; 21: 5280–5293.
Wang X, Robbins J . Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol 2014; 71: 16–24.
Chesser AS, Pritchard SM, Johnson GW . Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front Neurol 2013; 4: 1–12.
Xilouri M, Brekk OR, Stefanis L . Alpha-synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol 2013; 47: 537–551.
Acknowledgements
We thank Michael Molstad, Yoon Jee Lee, Keum Young Kang, Suhyun Lee, Soo Young Min and Sung Tae Kim for editorial help. This work was supported by the Seoul National University Nobel Laureates Invitation Program (to AC), the Basic Science Research Program (NRF-2013R1A2A2A01014170 to YTK) of the National Research Foundation (NRF) funded by the Ministry of Science, ICT and Future Planning (MSIP) of Korea, NIH grant HL083365 (to YTK and Song Li), the Dr Miriam and Sheldon G Adelson Medical Research Foundation (AMRF), the Israel Science Foundation (ISF), the I-CORE Program of the Planning and Budgeting Committee and the ISF (Grant1775/12), the EU Treat PolyQ Network, and the Deutsch-Israelische Projektkooperation (DIP). AC is an Israel Cancer Research Fund (ICRF) USA Professor.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article

Cite this article
Ciechanover, A., Kwon, Y. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47, e147 (2015). https://doi.org/10.1038/emm.2014.117
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2014.117
This article is cited by
-
Glial senescence enhances α-synuclein pathology owing to its insufficient clearance caused by autophagy dysfunction
Cell Death Discovery (2024)
-
Targeted degradation of ⍺-synuclein aggregates in Parkinson’s disease using the AUTOTAC technology
Molecular Neurodegeneration (2023)
-
P2X7 receptor inhibition ameliorates ubiquitin–proteasome system dysfunction associated with Alzheimer’s disease
Alzheimer's Research & Therapy (2023)
-
Recognition of an Ala-rich C-degron by the E3 ligase Pirh2
Nature Communications (2023)
-
Levosimendan inhibits disulfide tau oligomerization and ameliorates tau pathology in TauP301L-BiFC mice
Experimental & Molecular Medicine (2023)
