
Abstract
Parent-of-origin (PofO) effects, such as imprinting are a phenomenon where the effect of variants depends on parental origin. Conventional association studies assume that phenotypic effects are independent of parental origin, and are thus severely underpowered to detect such non-Mendelian effects. Risk of orofacial clefts is influenced by genetic and environmental effects, the latter including maternal-specific factors such as perinatal smoking and folate intake. To identify variants showing PofO effects in orofacial clefts we have used a modification of the family-based transmission disequilibrium test to screen for biased transmission from mothers and fathers to affected offspring, biased ratios of maternal versus paternal transmission, and biased frequencies of reciprocal classes of heterozygotes among offspring. We applied these methods to analyze published genome-wide single-nucleotide polymorphism (SNP) data from ∼2500 trios mainly of European and Asian ethnicity with non-syndromic orofacial clefts, followed by analysis of 64 candidate SNPs in a replication cohort of ∼1200 trios of European origin. In our combined analysis, we did not identify any SNPs achieving conventional genome-wide significance (P<5 × 10−8). However, we observed an overall excess of loci showing maternal versus paternal transmission bias (P=0.013), and identified two loci that showed nominally significant effects in the same direction in both the discovery and replication cohorts, raising the potential for PofO effects. These include a possible maternal-specific transmission bias associated with rs12543318 at 8q21.3, a locus identified in a recent meta-analysis of non-syndromic cleft (maternal-specific P=1.5 × 10−7, paternal-specific P=0.17). Overall, we conclude from this analysis that there are subtle hints of PofO effects in orofacial clefting.
Similar content being viewed by others

Introduction
Orofacial clefting (OFC) is one of the most common of all human congenital birth defects with a prevalence rate of 1 in 700 live births worldwide.1, 2 It may either occur in the context of malformation syndromes, or as an isolated anomaly (ie, non-syndromic OFC, NSOFC). Non-syndromic clefts are divided into two categories on the basis of epidemiological and embryological research findings: (i) cleft lip with or without cleft palate (CL/P) and (ii) cleft palate only (CPO).3, 4
Although genome-wide association studies (GWAS) have identified several genetic loci associated with risk oral clefting,5, 6, 7 there is significant evidence of other biological factors contributing to etiology of OFC.8 These may include parent-of-origin (PofO) effects such as imprinting,9 and other effects such as maternal–fetal interactions. In fact, environmental studies have indicated maternal smoking, and potentially alcohol consumption, during pregnancy greatly increase the risk of clefting in offspring, with some evidence of genetic interactions.2, 10, 11 Similarly, according to several studies, multi-vitamin supplements with or without folic acid taken during pregnancy have been shown to decrease the risk of oral clefting, with a stronger effect seen in CL/P as compared with CPO.12, 13, 14 If levels of circulating folate are influenced by genetic factors, then it can be hypothesized any maternal genes involved in dictating circulating folate levels could also alter the risk of OFCs in the fetus. Indeed, evidence in support of this idea has recently been published.15 As such, some genetic effects on disease risk can operate via a mechanism of maternal/fetal interaction. Previous candidate gene studies have suggested evidence for PofO effects in CL/P providing a strong rationale for performing comprehensive analysis to identify unconventional modes of inheritance.16, 17, 18, 19
However, non-Mendelian inheritance patterns such as imprinting are generally ignored in conventional GWAS, as these tests ignore the differential effects of maternally and paternally inherited alleles on phenotype. Indeed, simple case–control GWAS are unable to address this question. A detailed assessment of the extent of PofO effects across the genome is therefore important for the proper understanding of genome function in relation to disease. Although individual candidate gene studies have suggested possible PofO effects in OFC, a recent GWAS specifically investigated the role of PofO effects in OFCs, but was unable to identify any loci showing genome-wide significant effects.20
Previously, we used association methods specifically designed to detect PofO effects, and applied these to successfully detect quantitative trait loci exerting PofO effects on the expression of imprinted genes missed by conventional approaches.21 These methods take advantage of trio data to first define the parental origin of each allele in the offspring based on rules of Mendelian inheritance, and then perform separate association analyses of the two parental genotypes.
To analyze the role of PofO effects in common complex diseases, here we have employed an extension of the transmission asymmetry test and parental asymmetry test, to detect PofO effects using single-nucleotide polymorphism (SNP) genotyping in trios.22, 23 We applied these methods to re-analyze publicly available genotype data from trios with NSOFC from the database of Genotypes and Phenotypes.24 Our strategy analyzes associations separately for the maternally and paternally derived alleles, providing considerably increased power to detect PofO effects over conventional GWAS approaches. By first annotating parental origin of each allele in the affected offspring, we can conduct specific tests for association with maternal and paternal alleles independently, together with additional tests to look for differential effects of alleles inherited maternally versus paternally. From this initial genome-wide screen, we then selected 64 SNPs showing the strongest putative transmission bias for follow-up in a replication cohort of ∼1200 additional trios of European origin. Although we failed to identify any loci with PofO affects achieving genome-wide significance, our results do provide evidence suggesting biases for maternally inherited genetic factors influencing the risk of NSOFC.
Methods
Genome-wide detection of putative PofO effects
Genome-wide SNP data for 7018 individuals comprising 2339 trios in which each child was affected with any type of NSOFC (CL/P or CPO), were downloaded from dbGaP (Accession number: phs000094.v1.p1).7 Available genotype data included a total of 1 387 466 SNPs, comprising 601 273 genotyped SNPs and an additional 786 193 SNPs with discrete genotypes imputed by BEAGLE using HapMap Phase II samples as a reference panel. After converting high confidence imputed SNPs at r2≥0.9 to their respective genotypes and filtering non-informative SNPs (see Supplementary Information), we identified the transmitted and non-transmitted alleles in each parent, and the paternally and maternally inherited alleles in each child using rules of Mendelian inheritance. We then performed four different tests to detect putative PofO effects: (i) analysis of transmission bias from heterozygous fathers to affected children (PAT); (ii) analysis of transmission bias from heterozygous mothers to affected children (MAT); (iii) a comparison with the maternal and paternal odds ratios (PofO); and (iv) a comparison of the relative frequency of the two classes of heterozygotes in affected children (HET) (Figure 1). We analyzed this data set using seven different combinations based on disease subtype (NSCL/P and NSCPO3, 4) and ethnic groups (Europeans and Asians). The number of SNPs and samples used in each analysis are shown in Table 1.

Summary of the method used to screen for parent-of-origin effects in orofacial clefts.
SNPs showing putative parental-specific transmission bias in this discovery cohort were defined as follows: as a primary filter, we first selected those SNPs showing nominal significance (PPofO<0.05) in the PofO test, despite the large number of tests. Then, using a significance threshold of P<1 × 10−5, SNPs were considered to show a possible PofO bias if they were significant in any of the four tests: PAT, MAT, PofO and HET. A subset of these SNPs was then carried forward for further investigation in a replication cohort (see below).
Using the PLINK -blocks function,25 we calculated the number of distinct linkage disequilibrium (LD) blocks containing GWAS SNPs showing PPofO<0.05, and either PMAT<10−4 or PPAT<10−4. Enrichment analysis was performed using χ2-test with d.f.=1, under the null hypothesis that there should be equal numbers of MAT and PAT LD blocks.
Replication study
We selected 64 SNPs (32 SNPs for NSOFC, 33 SNPs for NSCL/P and 5 SNPs for NSCPO; with six SNPs shared between NSOFC and NSCL/P) for a replication study using 1197 European trios. Seven hundred and forty six trios were part of the EUROCRAN/ITALCLEFT studies (273 from the Netherlands, 124 from Italy, 118 from the UK, 73 from Slovakia, 71 from Hungary, 33 from Bulgaria, 23 from Slovenia, 21 from Estonia and 10 from Spain), and the 451 remaining trios were recruited in Bonn, Germany.6 Two additional SNPs overlapping SEMA4D, a gene with roles in axon guidance,26 were also included for CLP+CPO replication analysis. At the phenotypic level, the sample was subdivided into 931 trios where the index patient had NSCL/P and 266 with NSCPO (see Supplementary Information).
Genotyping was conducted using Sequenom MALDI-ToF mass spectrometer MassArray system (Sequenom Inc., San Diego, CA, USA). Primers were synthesized at Metabion (Martinsried, Germany). Using Sequenom MassARRAY Assay Design Software 3.4, two multiplex assays comprising all 64 selected SNPs plus the gender-specific variant were designed. Genotype data were analyzed using Sequenom Spectrodesigner Software package. Inter- and intraplate duplicates were included to check for genotype consistencies across DNA plates. Allele peaks were analyzed using Sequenom Typer Analysis software and genotype calls were confirmed by visual inspection of cluster plots. After quality control filtering, our final filtered replication data set comprised 48 SNPs mapping to 38 distinct chromosomal loci (see Supplementary Information). As the patient consent does not allow any unrestricted release of data, even in anonymised form, genotypes were not deposited in a public database. However, genotypes can be provided by the authors upon request.
In the replication sample PAT, MAT, PofO and HET tests for analysis of PofO effects were performed, as described above. Subsequently, combined analyses in which we pooled both the discovery and replication samples were conducted.
Results
Genome-wide analysis of PofO effects
Table 1 summarizes the results for genome-wide analysis of PofO effects in the different phenotypic and ethnic groups analyzed. Based on a low stringency discovery threshold of P<1 × 10−5 in any of the four tests for PofO effects (PAT, MAT, PofO and HET), among the combined NSCL/P and NSCPO samples, we identified a total of 55 SNPs (representing 13 distinct LD blocks) when the two ethnic groups were analyzed together, 31 SNPs (21 LD blocks) using only European samples, and 9 SNPs (8 LD blocks) using only Asian samples (Supplementary Table 1). Similarly, when performing our analysis based on each disease subtype, we identified an additional 52, 21 and 16 SNPs (falling in 14, 17 and 12 separate LD blocks) associated with NSCL/P based on the analysis of all samples combined (Europeans and Asians), respectively (Supplementary Table 2). In the analysis of NSCPO, we identified 36 SNPs (representing 19 LD blocks) when analyzing all ethnic groups (Supplementary Table 3).
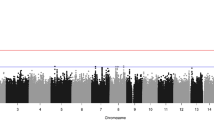
The most significant site, showing a putative association only when transmitted by the mother, was produced by SNP rs3814878 (16p11.2) in the MAT test (PMAT=1.69 × 10−7). This same SNP showed no significant transmission bias in PAT test (PPAT=0.052) (Figure 2, SNP M11). This region contains an interesting candidate gene for OFC, namely TBX6 (Supplementary Figure 1).

Manhattan plot showing the results of four tests for 22 autosomes performed using all ethnic groups (Europeans and Asians) and disease subtypes (CL/P and CPO) combined. The y axis shows the −log10 P-value for each test and the red and blue lines represent thresholds of P<1 × 10−5 (suggestive association) and 0.05 (nominal significance), respectively. Green dots represent loci showing suggestive associations identified by the four tests (see Methods). Each locus is labeled based on the test showing P<1 × 10−5: P# for PAT, M# for MAT, PofO# for PofO test and H# for HET. The P-values generated by four tests for each locus are shown in all four panels using the same label. For example, all SNPs at M11 loci have P<10−5 in MAT (above red line), while the same SNPs show P>0.05 in PAT (below blue line). These SNPs show nominal significance in PofO and HET (between blue and red line).
To investigate whether there was any bias in the transmission ratio of nominally significant associations between the two parental alleles, we calculated the number of independent loci (LD blocks) containing any SNP exceeding a threshold of P<1 × 10−4 in the MAT and PAT tests for each ethnic group and each category of OFC (Figure 3). In every case the total number of maternal-specific signals equaled or exceeded the number of paternal-specific signals, with a significant excess of maternal-specific signals observed in three categories: All (NSCL/P+NSCPO, P=0.013), Europeans (NSCL/P+NSCPO, P=0.026) and All (NSCPO, P=0.003). A similar excess of loci showing maternal-specific transmission bias was also observed using SNPs with an increased significance threshold of P<1 × 10−5 in the MAT and PAT tests, although due to the smaller number of loci, these differences did not reach statistical significance (data not shown).

A global bias for maternal genetic effects in the etiology of OFCs. In every comparison using different subtypes of orofacial clefts and ethnicities (All: all ethnic groups, Eur: Europeans and Asn: Asians), we observed that the number of maternal-specific signals equals or exceeds the number of paternal-specific signals. A significant excess (P<0.05) of maternal-specific signals is observed in three categories, as indicated. Bar plots show the number of independent loci (LD blocks) with P<1 × 10−4 identified using the MAT (gray) and PAT (black) tests. A similar excess of maternal associations was also observed using a more stringent significance threshold of P<10−5, although due to the smaller number of loci, these differences did not reach statistical significance (data not shown).
We calculated the genomic inflation factors for the MAT and PAT tests (Supplementary Table 4). We observed a small increase in the genomic inflation factor for the MAT compared with the PAT in all categories, although the magnitude of this bias is small, with the maximum genomic inflation factor observed being 1.04. QQ plots for the MAT and PAT are shown in Supplementary Figure 2.
Replication study
The results of replication analysis using 48 SNPs passing all quality filtering steps are summarized in Table 2 and Supplementary Table 5.
Among the SNPs exceeding our low stringency discovery thresholds in the initial GWAS (P≤1 × 10−5), only two passed our thresholds for successful replication, showing P≤0.01 with the MAT or PAT in the replication cohort in the same direction as observed in the discovery phase. Considering the replication data alone, the most significant P-value observed was PPAT=0.002 at SNP rs719325 in NSCL/P (Figure 4). This same SNP yielded PPAT=8.1 × 10−6 in the discovery GWAS (using combined European and Asian samples), giving a combined PPAT=5.4 × 10−8. However, although in the discovery GWAS this SNP yielded PPofO=0.005 and PMAT=0.69 suggesting this locus shows a paternal-specific transmission bias, this effect was not seen in the replication cohort, with PPofO=0.41 and PMAT=0.048. Results using a conventional TDT test for this SNP were PTDT=7.8 × 10−4 in the discovery GWAS, PTDT=0.002 in the replication cohort, yielding a combined P=4.9 × 10−6. A second SNP rs12543318, yielded PMAT=2.2 × 10−6 in the discovery GWAS (NSOFC in European samples), and PMAT=0.004 in the replication cohort, giving a combined PMAT=1.5 × 10−7 (Figure 4). However, although this SNP yielded PPofO=0.009 in the GWAS PofO test, and PPAT=0.24 and 0.43 in the discovery and replication cohorts, respectively, suggesting a putative maternal-specific transmission bias, results for the PofO test in the replication cohort were not clear (P=0.12). Results using a conventional TDT test for this SNP were PTDT=1.2 × 10−4 in the discovery GWAS, PTDT=0.022 in the replication cohort, yielding a combined PTDT=2.4 × 10−5. This same SNP also yielded very similar results in NSCL/P using both European alone, and the combined European and Asian GWAS and replication samples.

Loci on 2q35 and 8q21.3 show suggestive evidence of PofO effects in NSCL/P in both discovery and replication cohorts. (a) A paternal-specific signal at SNP rs719325 on chromosome 2q35 in combined European and Asian analysis (PGWAS: PPAT=8.07 × 10−6, PMAT=0.69, Pcomb: PPAT=5.43 × 10−8, PMAT=0.12). (b) A maternal-specific signal at SNP rs12543318 on chromosome 8q21.3 in European-only analysis (PGWAS: PPAT=0.31, PMAT=2.89 × 10−6, Pcomb: PPAT=0.27, PMAT=7.85 × 10−7). The squares and diamond represent P-values from the discovery GWAS and combined analysis, respectively.
Results for the 48 SNPs using a conventional TDT (which does not consider parental origin) are listed in Supplementary Table 6. We observed four SNPs (corresponding to three LD blocks) yielding both PTDT<0.01 in the discovery cohort and PTDT<0.05 in the replication cohort.
Discussion
In this study, we have applied modified versions of the Transmission Asymmetry Test and Parental Asymmetry Test to perform genome-wide screening for PofO effects associated with non-syndromic oral cleft in mother/father/affected child trios. We used a previously published genome-wide SNP data from ∼2500 trios for discovery,7 followed by genotyping of selected SNPs in a replication cohort of ∼1200 trios. Using four tests (PAT, MAT, PofO and HET – see Methods), a low stringency threshold in the discovery cohort identified a total of 210 SNPs (corresponding to 88 independent regions) showing evidence of possible parental-specific transmission bias associated with risk of non-syndromic oral clefts (NSCL/P or NSCPO) in various ethnic groups (all, European or Asian populations). Of these discovery SNPs, 64 were carried forward for testing in the replication cohort.
Although in our replication analysis we observed several loci reaching nominal significance in the same direction as the discovery GWAS data, none of the markers tested achieved generally accepted definitions of genome-wide significance (P<5 × 10−8) in the combined sample. However, there are several possible explanations for failure to replicate GWAS signals, including the ‘winners curse’ phenomenon.27 Of particular note, our replication cohort was approximately half the size of our discovery cohort, and as a result is underpowered to detect small effects on disease risk detected in our discovery phase.
We identified two loci that passed our thresholds for successful replication (P≤1 × 10−5 in the discovery cohort and P≤0.01 in replication with either the MAT or PAT), raising the potential for PofO effects tagged by these loci. Paternally biased transmission was detected for rs719325 at 2q25, and this result approached genome-wide significance in the combined analysis (PPAT=5.4 × 10−8). This marker is located ∼164 kb upstream of SLC4A3, a gene which encodes a trans-membrane transport protein involved in regulation of intracellular pH. SLC4A3 has been described as a candidate gene for human retinal degeneration,28 however, no role in craniofacial development has been described so far. Aside from genomic imprinting, mechanistically it is harder to conceptualize how paternal-specific effects on disease risk might operate, although some have been recognized.9
We also identified a possible maternal-specific transmission bias associated with rs12543318 located within 8q21.3. This same SNP was recently identified as a susceptibility locus for NSCL/P in a recent meta-analysis.29 This marker maps to an intergenic region for which, so far, no functional information related to OFC is available. Although the meta-analysis of Ludwig et al.29 and the current study utilized many of the same individuals, this previous study did not test for PofO effects. Comparison of the TDT results for the NSCL/P group in the present and the trio-part of Ludwig et al29 reveals they are in the same range (with the slight difference being attributed to different quality controls on samples). However, the present PofO analysis suggests the signal at rs12543318 is largely attributable to maternally derived alleles, as our MAT test yielded a combined P=1.5 × 10−7, while the corresponding PAT test gave a combined P=0.17, with a combined PPofO=0.004. No known imprinting effects map to this region. Thus, the 8q21.3 region might contain a genetic element which in some way interacts with other maternal-specific risk effects.
Two other loci that achieved suggestive evidence of replication (P≤1 × 10−5 in discovery and P≤0.1 in replication with the MAT) are worthy of mention. rs17447439 maps within an intron of the TP63 gene at 3q28, and showed weak evidence of a maternal-specific transmission bias. Several studies provide evidence linking TP63 with the development of OFC. For example, a homozygous mutation in TP63 has been suggested to have a causative role in NSCL/P.30 Further, a recent genome-wide analysis of p63-binding sites identified the AP-2α transcription factor as target site.31 AP-2α is known to be essential for craniofacial development and cranial closure32 and has been implemented in NSCL/P.33 Similarly in the analysis of NSCPO, a suggestive bias for maternal over-transmission was observed for rs6539608 located on chromosome 12q21. No genes or transcripts map to this region, making an interpretation of functional relevance difficult. Notably, however, Koillinen et al34 found suggestive linkage to this locus in a study comprising nine Finnish multiplex families affected with NSCPO.34
On balance our analysis finds only weak evidence for specific genetic loci individually contributing to PofO effects in OFC. After performing replication analysis of 48 SNPs showing suggestive evidence of PofO effects in the discovery cohort, individually none of these showed unambiguous confirmation of statistical signals that would allow us to define any PofO effects with confidence. These results are broadly consistent with the study of Shi et al,20 which utilized an alternative method to analyze the same discovery cohort. One of the limitations of our method compared with the method used in Shi et al20 is the inability to differentiate between different types of parental biases, such as imprinting or maternal effects. Our analysis approach also does not directly consider environmental effects such as the maternal intrauterine environment. One advantage of our method, however, is that by performing independent tests of the maternally and paternally derived alleles (MAT and PAT tests) we are able to directly compare the relative influence of the two parental genomes on risk of OFCs in the child. Our stratified analyses by ethnic groups and type of OFC showed the number of loci showing a maternal-specific transmission bias equaled or exceeded the number of paternal-specific signals, with a significant excess of maternal-specific signals observed in three categories. Although this approach cannot discern the underlying mechanism, this observation suggests overall maternally inherited alleles exert a more significant effect on the risk of OFCs in offspring than do paternally derived alleles. In this regard, it is interesting to note effects of maternal genotype and maternal-specific environmental modifiers such as smoking, drinking and vitamin intake during pregnancy on oral cleft susceptibility have been previously reported in various studies.2, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 However, it should be noted that the parental asymmetry tests we applied here do not specifically test for maternal effects which would be expected to occur via alterations of the in utero environment. Calculation of genomic inflation factors for the MAT and PAT tests showed a small increase for the MAT compared with the PAT in all categories, this increased inflation factor in the MAT does not discern the underlying cause, being consistent with either a true excess of biological associations with the maternally inherited alleles, or alternatively differing population structure between the two parental populations.35 In addition, comparison of the 64 SNPs used in replication analysis with two online catalogs of known imprinted loci (http://igc.otago.ac.nz/home.html and http://www.geneimprint.com/) showed that none are located within 1 Mb of any known imprinted loci. However, we do note that one study recently reported TBX6, the most significant locus identified in our genome-wide screen for PofO effects in OFCs, as showing evidence of imprinting in placental tissue.36 As a result, we suggest that further studies of this locus in OFCs are warranted.
Although we utilized four different tests (MAT, PAT, PofO and HET), we did not apply any multiple testing correction for several reasons. First, as the PofO and HET tests share the same underlying data used by the MAT and PAT these four tests are not independent. Second, although we utilized a combined significance threshold of P<5 × 10−8, our primary filter in the discovery cohort was to select SNPs showing PPofO<0.05, and thus the MAT and PAT tests were only applied to a relatively small fraction of the total genotypes available. Third, the use of a two-stage discovery and replication design in an additional cohort of ∼1200 trios of European descent, should be considerably more robust than any previous studies of PofO effects in OFC.
Both the method we have used, and that used by Shi et al20 are heavily dependent on SNPs that tag loci showing PofO-specific transmission biases. Using this assumption, we can identify some types of PofO effects, but not all. We also analyzed our discovery and replication data using a conventional TDT which does not consider parental origin. In this analysis, we observed three loci yielding nominal replication with both PTDT<0.01 in the discovery cohort and PTDT<0.05 in the replication cohort (combined discovery plus replication TDT P-values ranging from 2.4 × 10−4 to 3.3 × 10−7). It should be noted that our MAT and PAT tests are essentially a modification of the TDT in which only one half of the genotypes inherited by each child are considered. When considered as individual tests, they are also capable of detecting risk alleles that do not exhibit PofO effects, although with reduced power compared with the conventional TDT. For example, as can be seen in Figure 2, previously identified OFC risk loci such as the 8q24 locus5 were detected, but as these show significant P-values in both the MAT and PAT tests strong PofO effects at these markers can be excluded. Therefore, a reasonable alternative interpretation of our results is that some or all of the loci we detected with suggestive PofO effects might simply represent weak risk loci for OFC, and which manifest in our data in only one of the two parental tests by chance due to insufficient power to reliably detect these effects in both MAT and PAT tests. As such, we suggest some of the loci we identify represent interesting candidate regions for future studies of OFC.
In conclusion, our study provides suggestive evidence for PofO effects in susceptibility to OFC, identifying several loci showing a parental transmission bias, and an overall excess of maternal-specific association signals in the genome. Given abundant evidence supporting a role for non-Mendelian and transgenerational inheritance patterns in a variety of different diseases and conditions,9 we suggest similar analyses considering parental origin of risk alleles will likely reveal novel PofO effects contributing to many human phenotypes.
Accession codes
References
Mossey PA, Little J : Epidemiology of oral clefts: an international perspective; In Wyszynski DF, (ed Cleft Lip and Palate): From Origin to Treatment 2002; 127–144.
Murray JC : Gene/environment causes of cleft lip and/or palate. Clin Genet 2002; 61: 248–256.
Lacheretz M, Poupard B : Inheritance of harelip and cleft palate. Reexamination apropos of statistics of 879 cases, 212 of them familial. Chirurgie 1972; 98: 264–270.
Sperber GH : Formation of the primary and secondary palate. Cleft Lip and Palate: From Origin to Treatment Oxford University Press 2002, pp 5–13.
Birnbaum S, Ludwig KU, Reutter H et al: Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet 2009; 41: 473–477.
Mangold E, Ludwig KU, Birnbaum S et al: Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat Genet 2010; 42: 24–26.
Beaty TH, Murray JC, Marazita ML et al: A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet 2010; 42: 525–529.
Mangold E, Ludwig KU, Nothen MM : Breakthroughs in the genetics of orofacial clefting. Trends Mol Med 2011; 17: 725–733.
Guilmatre A, Sharp AJ : Parent of origin effects. Clin Genet 2011; 81: 201–209.
Krapels IP, Raijmakers-Eichhorn J, Peters WH, Roelofs HM, Ras F, Steegers-Theunissen RP, Eurocran Gene-Environment Interaction Group: The I,105V polymorphism in glutathione S-transferase P1, parental smoking and the risk for nonsyndromic cleft lip with or without cleft palate. Eur J Hum Genet 2008; 16: 358–366.
van den Boogaard MJ, de Costa D, Krapels IP et al: The MSX1 allele 4 homozygous child exposed to smoking at periconception is most sensitive in developing nonsyndromic orofacial clefts. Hum Genet 2008; 124: 525–534.
Badovinac RL, Werler MM, Williams PL, Kelsey KT, Hayes C : Folic acid-containing supplement consumption during pregnancy and risk for oral clefts: a meta-analysis. Birth Defects Res A Clin Mol Teratol 2007; 79: 8–15.
Wilcox AJ, Lie RT, Solvoll K et al: Folic acid supplements and risk of facial clefts: national population based case-control study. BMJ 2007; 334: 464.
Johnson CY, Little J : Folate intake, markers or folate status and oral clefts: is the evidence converging? Int J Epidemiol 2008; 37: 1041–1058.
Bufalino A, Ribeiro Paranaiba LM, Nascimento de Aquino S, Martelli-Junior H, Oliveira Swerts MS, Coletta RD : Maternal polymorphisms in folic acid metabolic genes are associated with nonsyndromic cleft lip and/or palate in the Brazilian population. Birth Defects Res A Clin Mol Teratol 2010; 88: 980–986.
Scapoli L, Martinelli M, Pezzetti F et al: Linkage disequilibrium between GABRB3 gene and nonsyndromic familial cleft lip with or without cleft palate. Hum Genet 2002; 110: 15–20.
Reutter H, Birnbaum S, Mende M et al: TGFB3 displays parent-of-origin effects among central Europeans with nonsyndromic cleft lip and palate. J Hum Genet 2008; 53: 656–661.
Sull JW, Liang KY, Hetmanski JB et al: Maternal transmission effects of the PAX genes among cleft case-parent trios from four populations. Eur J Hum Genet 2009; 17: 831–839.
Sull JW, Liang KY, Hetmanski JB et al: Excess maternal transmission of markers in TCOF1 among cleft palate case-parent trios from three populations. Am J Med Genet A 2008; 146A: 2327–2331.
Shi M, Murray JC, Marazita ML et al: Genome wide study of maternal and parent-of-origin effects on the etiology of orofacial clefts. Am J Med Genet A 2012; 158A: 784–794.
Garg P, Borel C, Sharp AJ : Detection of parent-of-origin specific expression quantitative trait loci by cis-association analysis of gene expression in trios. PLoS One 2012; 7: e41695.
Weinberg CR, Wilcox AJ, Lie RT : A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet 1998; 62: 969–978.
Weinberg CR : Methods for detection of parent-of-origin effects in genetic studies of case-parents triads. Am J Hum Genet 1999; 65: 229–235.
Mailman MD, Feolo M, Jin Y et al: The NCBI dbGaP database of genotypes and phenotypes. Nat Genet 2007; 39: 1181–1186.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Isbister CM, Tsai A, Wong ST, Kolodkin AL, O‘Connor TP : Discrete roles for secreted and transmembrane semaphorins in neuronal growth cone guidance in vivo. Development 1999; 126: 2007–2019.
Xiao R, Boehnke M : Quantifying and correcting for the winner’s curse in genetic association studies. Genet Epidemiol 2009; 33: 453–462.
Downs LM, Wallin-Hakansson B, Boursnell M et al: A frameshift mutation in golden retriever dogs with progressive retinal atrophy endorses SLC4A3 as a candidate gene for human retinal degenerations. PLoS One 2011; 6: e21452.
Ludwig KU, Mangold E, Herms S et al: Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet 2012; 44: 968–971.
Leoyklang P, Siriwan P, Shotelersuk V : A mutation of the p63 gene in non-syndromic cleft lip. J Med Genet 2006; 43: e28.
McDade SS, Henry AE, Pivato GP et al: Genome-wide analysis of p63 binding sites identifies AP-2 factors as co-regulators of epidermal differentiation. Nucleic Acids Res 2012; 40: 7190–7206.
Schorle H, Meier P, Buchert M, Jaenisch R, Mitchell PJ : Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature 1996; 381: 235–238.
Rahimov F, Marazita ML, Visel A et al: Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet 2008; 40: 1341–1347.
Koillinen H, Lahermo P, Rautio J, Hukki J, Peyrard-Janvid M, Kere J : A genome-wide scan of non-syndromic cleft palate only (CPO) in Finnish multiplex families. J Med Genet 2005; 42: 177–184.
Yang J, Weedon MN, Purcell S et al: Genomic inflation factors under polygenic inheritance. Eur J Hum Genet 2011; 19: 807–812.
Yuen RK, Jiang R, Peñaherrera MS et al: Genome-wide mapping of imprinted differentially methylated regions by DNA methylation profiling of human placentas from triploidies. Epigenetics Chromatin 2011; 4: 10.
Acknowledgements
We thank Shaun Purcell for helpful discussions. This work was supported by NIH Grants HD073731, DA033660, HG006696 and Grant NIRG69983 from the Alzheimer’s Association to AJS, and also by the Deutsche Forschungsgemeinschaft (FOR 423 and individual Grants MA 2546/3-1, KR 1912/7-1, NO 246/6-1, WI 1555/5-1). KUL and ACB received support from the BONFOR program of the Medical Faculty, University of Bonn. The details of the collection and methods for samples used in this study are described by Beaty et al.7 The data sets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000094.v1.p1 through dbGaP accession number phs000094.v1.p1. Funding support for the study entitled ‘International Consortium to Identify Genes and Interactions Controlling Oral Clefts’ was provided by several previous Grants from the National Institute of Dental and Craniofacial Research (NIDCR), including: R21-DE-013707, R01-DE-014581, R37-DE-08559, P50-DE-016215, R01-DE-09886, R01-DE-012472, R01-DE-014677, R01-DE-016148, R21-DE-016930; R01-DE-013939. Additional support was provided in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences, the Smile Train Foundation for recruitment in China and a Grant from the Korean government. The genome-wide association study, also known the Cleft Consortium, is part of the Gene Environment Association Studies (GENEVA) program of the trans-NIH Genes, Environment and Health Initiative [GEI] supported by U01-DE-018993. Genotyping services were provided by the Center for Inherited Disease Research (CIDR), funded through a federal contract from the National Institutes of Health (NIH) to The Johns Hopkins University, contract number HHSN268200782096C. Assistance with genotype cleaning, as well as with general study coordination, was provided by the GENEVA Coordinating Center (U01-HG-004446) and by the National Center for Biotechnology Information (NCBI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Garg, P., Ludwig, K., Böhmer, A. et al. Genome-wide analysis of parent-of-origin effects in non-syndromic orofacial clefts. Eur J Hum Genet 22, 822–830 (2014). https://doi.org/10.1038/ejhg.2013.235
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.235

{kind=link}
{kind=link}