
Abstract
Mandibuloacral dysplasia (MAD) is a rare disease resulting from a mutation of LMNA gene encoding lamins A and C. The most common mutation associated with this disease is a homozygous arginine 527 replacement by histidine. Three female patients originating from two unrelated families from Northeast Egypt were examined. Their growth was retarded; they had microcephaly, widened cranial sutures, prominent eyes and cheeks, micrognathia, dental crowding, hypoplastic mandible, acro-osteolysis of distal phalanges, and joint contractures. In addition, they presented some progeroid features, such as pinched nose, premature loss of teeth, loss of hair, scleroderma-like skin atrophy, spine rigidity, and waddling gait. The clinical presentation of the disease varied between the patient originating from Family 1 and patients from Family 2, suggesting that unknown, possibly epigenetic factors, modify the course of the disease. The first symptoms of the disease appeared at the age of 2.5 (a girl from Family 1), 5, and 3 years (girls from Family 2). All patients had the same, novel homozygous c.1580G>T LMNA mutation, resulting in the replacement of arginine 527 by leucine. Computational predictions of such substitution effects suggested that it might alter protein stability and increase the tendency for protein aggregation, and as a result, might influence its interaction with other proteins. In addition, restriction fragment-length polymorphism analysis performed in 178 unrelated individuals showed that up to 1.12% of inhabitants of Northeast Egypt might be heterozygous carriers of this mutation, suggesting the presence of a founder effect in this area.
Similar content being viewed by others

Introduction
Mandibuloacral dysplasia type A (MAD type A (MADA); OMIM # 248370) is a rare disease with a recessive trait of inheritance. Its most common features are: postnatal growth retardation, skeletal abnormalities such as hypoplasia of the mandible and of clavicles, acro-osteolysis of the terminal phalanges, delayed closure of the cranial sutures, dental overcrowding, joint contractures, skin atrophy with mottled pigmentation, lipodystrophy limited to extremities, as well as metabolic abnormalities such as insulin-resistance, diabetes, and hypertriglyceridemia. Affected individuals might also present progeroid features such as bird-like face with micrognathia, pinched nose, prominent eyes, hair loss, scleroderma-like skin changes, and nail dysplasia. MADA is associated with primary laminopathy resulting from mutation in the LMNA gene1 encoding lamin A and C, the main components of the nuclear lamina. In addition, MAD type B, in which lipodystrophy is generalized, results from a secondary laminopathy caused by mutations in ZMPSTE-24 gene encoding zinc metalloproteinase FACE-1 responsible for post-translational modification of prelamin A.2
The most common mutation reported in patients with MADA is the homozygous p.Arg527His substitution in exon 9 of the LMNA gene.1, 3, 4 Other LMNA mutations resulting in MAD are homozygous p.Ala529Val and compound p.Arg527His/Val440Met.5, 6 MAD with progeria-like features was described in relation to the homozygous p.Arg527Cys, homozygous p.Lys542Asn, homozygous p.Arg471Cys, compound p.Thr528Met/Met540Thr, and compound p.Arg471Cys/Arg527Cys LMNA mutations.7, 8, 9, 10, 11
In this work, we describe a novel homozygous p.Arg527Leu substitution mutation in the LMNA gene, resulting in MAD with progeroid features. We also suggest that a founder effect might be present in Northeast Egypt.
Subjects and methods
Subjects
Family 1: patient APS 1201
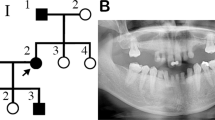
A 4-year-old female patient (APS 1201, Figure 1) was referred to the Genetics Unit, Mansoura University Children’s Hospital (MUCH) in Mansoura, Egypt, because of frequent episodes of obstructive sleep apnea with exacerbations of upper airway infections. She was born to unrelated parents (Figure 2a); however, both parents originated from families living in the same small village of Salamon Al-Komash, Dakahlia Governorate, and a distant kinship cannot be completely ruled out.

Current photographs of the affected children showing characteristic MAD and progeroid features.

(a) Pedigrees of the two families affected by a progeroid syndrome, showing autosomal recessive trait of inheritance. (b) Sequencing of exon 9 of LMNA revealed the presence of a homozygous c.1580 G>T mutation, resulting in the replacement of arginine by leucine at amino acid position 527 (p.Arg527Leu) – proband from Family 2. (c) Heterozygous mutation present in the mother of the probands from Family 2. (d) Wild-type alleles present in the unaffected grandfather from the same family.
Family 2: patient APS 2301 and patient APS 2302
Two sisters aged 10 and 4 years (APS 2301 and APS 2302, respectively, Figure 1), inhabitants of Ismalia city, Egypt, born to consanguineous parents (Figure 2a), were referred to the Genetics Unit, MUCH, Mansoura, Egypt, because of impaired motor skills (such as difficulty in walking) and to marked stiffness in the hands, feet, and back. Both had been born by a normal, vaginal delivery. At birth, they were completely normal, with average birth weight and height, good suckling, and fair neonatal reflexes.
Blood smear
Blood smears were routinely stained using May–Grunwald–Giemsa method.
DNA isolation
Genomic DNA was extracted from peripheral blood mononuclear cells using the Capture Column Kit (Qiagen, Hilden, Germany).
Genetic testing
A total of 12 exons of LMNA, 10 exons of ZMPSTE-24, and exon–intron boundaries were amplified12, 13 and sequenced with a Big Dye Terminator Sequencing Ready Reaction kit (Applied Biosystems, Carlsbad, CA, USA) on the ABI PRISM 373 fluorescent DNA sequencer (Applied Biosystems).
Restriction fragment length polymorphism
Exon 9 of the LMNA gene was amplified by PCR with forward 5′-TTGGGTGGCGATGGGAGCGC-3′ and reverse 5’-GCAGCTGGCTCCATGTTGG-3′ primers. The 282 bp PCR product was restricted with the CviQI enzyme (New England Biolabs, Ipswich, MA, USA). The wild-type allele was represented by 134 and 148 bp fragments, whereas the mutated allele remained unrestricted.
Bioinformatics analysis
We predicted the effects of amino acid substitution on protein stability and function on the basis of the following methods: PolyPhen, PMut, SNPeffect, SIFT, Mupro, PoPMuSiC, CUPSAT, HOPE, nsSNPAnalyzer, and SNPs&GO.14, 15, 16, 17, 18, 19, 20, 21, 22, 23 The crystal structure of the lamin immunoglobulin-like (IG-like) domain (pdb id: 1IFR) was used as an input wherever possible; otherwise, the protein sequence was used.
Results
Clinical presentation
Patient APS 1201 was a full-term, healthy female, born by a spontaneous vaginal delivery, with normal development milestones until the age of 2 years. The first symptoms of the disease appeared when she was 2.5 years old; since then, she has been frequently admitted to the General, Pulmonary, Cardiology, and ENT wards of MUCH, because of a chronic upper airway obstruction associated with exacerbations of upper airway infections. Initially, adenoid hypertrophy was suspected; however, ENT consultants raised the possibility of mandibular dysplasia that might require reconstructive surgery to relieve the airway obstruction and obstruction-related infections. Next, she developed symptoms suggestive of a right-sided heart failure. Cardiac examination revealed marked systemic congestion, pulsating neck veins and a congested tender liver with the typical precordial signs of pulmonary hypertension (right ventricular uplift, accentuated second heart sound with close splitting, ejection systolic with early diastolic murmurs). The electrocardiogram showed right ventricular hypertrophy with marked right-axis deviation. Echocardiography revealed intact inter-ventricular and inter-atrial septa, poor cardiac muscle performance with reduced ejection fraction, and marked elevation of pulmonary blood pressure (45 mmHg). Respiratory function tests were normal. A diagnosis of sub-acute cor pulmonale due to upper airway obstruction was made, and treatment with oxygen inhalation, diuretic therapy, and digitalis was successful.
Symptoms detected at age 4 are described in Table 1. Basic biochemical parameters and results of other tests are shown in Table 2. Notably, no metabolic abnormalities were present, most probably because of the young age of the patient. The patient remains under the control of the Cardiac Clinic; she is maintained on diuretics and load reducers. To relieve the upper airway compromise, until maxillofacial reconstructive surgery is performed, she receives (on an ‘on-demand’ basis) a continuous positive airway pressure breathing (nasal CPAP) using a portable device.
Patient APS 2301 developed difficulties in leaning forward, in the ability to perform fine movements of hands (such as feeding herself and writing), and in movements of feet, all being a result of marked joint stiffness, at age 5. Plain radiographs of her hands and chest showed marked osteolysis (acro-osteolysis of terminal phalanges and absent clavicles; Figures 3a and b, respectively). She attended the Orthopedic, Dermatology, and Rheumatology Clinics; however, regardless of the prescribed treatment, symptoms were not relieved and systematically progressed. Patient APS 2302 started to have similar symptoms at age 3. Neither girl had any symptoms related to the respiratory system, they were normotensive without dyslipidemia.

Clinical features in a 10-year-old girl from Family 2 (APS 2301). (a) Plain radiograph of hands showing a claw deformity and acro-osteolysis of terminal phalanges. (b) Plain radiograph of chest showing absence of clavicles. (c) Blood smear stained using May–Grunwald–Giemsa method ( × 500 magnification) showing normal erythrocytes, and a lymphocyte with irregularly shaped nucleus. (d) Lymphocytes with irregularly shaped nuclei with outpouching ( × 1000 magnification). Black arrow – lymphocyte with irregular nucleus. White arrow – granulocyte.
Symptoms observed in Patient APS 2301 at age 10 and in Patient APS 2302 at age 4 are listed in Table 1, whereas the results of biochemical and of other tests are shown in Table 2. No metabolic abnormalities were noticed. They currently receive palliative physical therapy to relieve joint stiffness. They also receive vitamins and adhere to a dietary regimen to prevent dyslipidemia.
Blood smear
As the parents of the affected children did not agree for a biopsy, we evaluated the patients’ blood smears. The majority (approximately 80% in the patient APS 2301) of the peripheral blood lymphocytes detected in routine blood smears stained with May–Grunwald–Giemsa method showed markedly irregular nuclear shape (Figures 3c and d).
Genetic testing
We first sequenced all exons and exon–intron boundaries of the LMNA and ZMPSTE-24 genes in the three affected children. In all of them, sequencing revealed the presence of a homozygous c.1580G>T point mutation in LMNA exon 9, which resulted in the replacement of arginine by leucine at position 527 (p.Arg527Leu) in the IG-like domain of lamin A (Figure 2b). Sequencing of ZMPSTE-24 revealed no mutations. Analysis of the sequence of the LMNA gene in the probands’ unaffected parents (Family 1: APS 1101 and APS 1102; Family 2: APS 2201 and APS 2202) revealed that all of them were heterozygous p.Arg527Leu mutation carriers (Figure 2c). In addition, identical heterozygous mutation was found in the unaffected grandmother (APS 2104), but not in the grandfather (APS 2103) of Family 2 (Figure 2d). Other grandparents were not available for analysis (Figure 2a).
As the same mutation has been found in two unrelated families, we decided to check its prevalence in Northeast Egypt, the region where both families live. Restriction fragment-length polymorphism analysis performed in 178 unrelated, randomly selected inhabitants of this region, showed that two of them were heterozygous carriers of the c.1580G>T LMNA mutation. The presence of this mutation was subsequently confirmed by sequencing. Therefore, approximately 1.12% of inhabitants of Northeast Egypt might be heterozygous carriers of the c.1580G>T-mutated LMNA allele, and the frequency of the mutated allele in this region is approximately 0.56%.
Computational predictions of the p.Arg527Leu LMNA substitution effects
Computational analysis showed that the p.Arg527Leu substitution leads to the disruption of a salt bridge, introduction of a hydrophobic residue on the protein surface, and to the formation of a cavity that increases the solvent-exposed hydrophobic surface (Figure 4). We predicted the effects of p.Arg527Leu substitution on protein stability and function on the basis of a series of algorithms (Table 3). Predictions returned by most methods agree that the substitution of the positively charged arginine to the non-charged, hydrophobic leucine, results in destabilization of the lamin A IG-like domain structure and may increase the protein aggregation tendency. The primary destabilization of the protein structure will cause secondary effects due to the alteration of lamin A interaction with its protein partners.

Cartoon representation of lamin A IG-like domain (pdb id: 1IFR). (a) Arginine 527 forms a salt bridge with glutamate 537 (residues shown in the space-filled representation, with carbon atoms shown in gray, nitrogen atoms shown in blue, and oxygen in red). This salt bridge stabilizes the β-sheet (β strands are shown as yellow arrows), and as a result, contributes to the stability of the entire IG-like domain fold. (b) The residues around the Arg527–Glu537 (R527–E537) pair are densely packed (all heavy atoms around the salt bridge are shown as gray balls). (c) The p.Arg527Leu substitution destroys the salt bridge, introduces a pocket on the protein structure, and exposes a hydrophobic leucine to the solvent.
Discussion
Mutations of the LMNA gene result in a wide range of diseases, termed ‘laminopathies’,24, 25 such as autosomal dominant and autosomal recessive Emery–Dreifuss muscular dystrophy (EDMD2 and EDMD3),12, 26 limb girdle muscular dystrophy type 1B,27 congenital muscular dystrophy,28 dilated cardiomyopathy 1A,29 Charcot-Marie-Tooth disorder type 2B,30 Dunningan-type familial partial lipodystrophy,31 MAD,1 Hutchinson–Gilford progeria,32, 33, 34 and restrictive dermopathy.13 Their phenotypes are quite diverse, most probably owing to the multiple molecular functions of lamin A, including the regulation of gene transcription, chromatin organization, DNA replication, maintenance of nuclear integrity, and mechanotransduction, that might be diversely affected by various mutations.35, 36
A difficulty arises when genotype–phenotype correlations are being made. In some laminopathies, there is a good correlation between the specific mutations of LMNA and the clinical manifestations of the disease; in others, multiple mutations present in distinct parts of LMNA might all result in a similar phenotype; in still other cases, the mutation of a single residue might result in a diverse phenotypes.37 Indeed, this was also true for our patients; even though they all had the same LMNA mutation, the symptomatology of their disease was not exactly the same. First, the initial, most pronounced symptoms of the disease in the girl from Family 1, were related to the respiratory tract and appeared at the age of 2.5 years, whereas the initial symptoms in girls from Family 2 were related to the stiffness of joints and appeared at the ages of 3 and 5. Second, the extent of typical MAD symptoms varied. For example, hypoplasia of the clavicle, joint contractures, and mottled hyperpigmentation were less evident in the 4-year-old girl from Family 1 than in the 4-year-old girl from Family 2 and in her older sister. Third, symptoms of progeria also differed between these girls: the child from Family 1 had neither high-pitched voice nor alopecia, whereas both symptoms were present in children from Family 2. Finally, acanthosis nigricans was present in children from Family 2, but absent in a child from Family 1. The difference in the extent of symptoms in the sisters from Family 2 might be explained by their age and by the time that has passed since the occurrence of the first symptoms. In contrast, differences in clinical manifestations of the disease between the child from Family 1 and children from Family 2 are not the result of age or time. We speculate that they are a result of diverse interactions of the mutated lamin A with other proteins or a result of different epigenetic modifications of the mutated LMNA.
Respiratory problems due to mandibular hypoplasia in MAD patients were not reported. However, they were observed in a child from Family 1; she suffered from obstructive sleep apnea complicated by pulmonary hypertension due to mandibular hypoplasia and backward displacement of tongue. Recently, pulmonary hypertension has been described in two patients suffering from classical progeria without cardiovascular impairment, diabetes mellitus, or hypertriglyceridemia. These patients, carriers of the BANF1 mutations, suffered from profound skeletal abnormalities and developed pulmonary hypertension due to severe scoliosis.38 In addition, patients with mandibular hypoplasia because of causes other than MAD (eg, fibrodysplasia ossificans progressiva) were reported to have obstructive sleep apnea causing significant pulmonary hypertension.39 It thus becomes plausible that other patients suffering from MAD might develop pulmonary hypertension at a later age, and therefore, should be carefully evaluated.
Our primary finding is the association of a novel homozygous c.1580G>T mutation of LMNA, resulting in the replacement of arginine 527 by leucine (p.Arg527Leu), with features of MADA and of progeria. The most common genetic cause of MADA is the homozygous p.Arg527His LMNA mutation.1, 3, 4 Furthermore, the homozygous p.Arg527Cys LMNA mutation results in early and severe MAD and progeria.7 According to the computational structure prediction, a basic arginine at position 527 of the wild-type lamin A, forms a salt bridge with the glutamate at position 537, thus stabilizing the structure of the conserved C-terminal IG-like domain of this protein.40, 41 Our computational structure prediction showed that the replacement of arginine 527 by a neutral, hydrophobic leucine, completely destroys the salt bridge, and this results in the destabilization of the IG-like domain structure and increases the tendency for protein aggregation. Such an alteration most likely affects the interactions of lamin A with other proteins, thereby influencing multiple cellular processes. This, in turn, might explain the severe clinical presentation of the disease observed in our patients: its early appearance and the presence of a full set of typical MADA symptoms together with some progeroid features. We also showed that the replacement of arginine 527 by a basic histidine results in the destabilization of the salt-bridge formation; however, it has less severe consequences than the effects predicted for the replacement with leucine. Finally, we showed that the replacement of arginine 527 with a neutral cysteine leads to a broad change in the local landscape. Moreover, cysteine at position 527 presents a tendency to form disulfide bridge with cysteine 522 (based on the disulfide bridge predictors DiANNA and DIpro,42, 43) which could affect protein folding. Importantly, results of our computational disease prediction correspond quite accurately with the phenotypes associated with mutations of amino acid 527: p.Arg527His that was predicted as the least pathogenic, results in MADA; p.Arg527Leu that was shown as intermediately pathogenic, results in MADA and progeria; finally, p.Arg527Cys that was predicted as the most pathogenic, results in an earliest and most severe MADA and progeria (Table 3).
The second major important finding described here is the common occurrence of the mutated allele in the inhabitants of Northeast Egypt; we found that almost 1.12% of them might be heterozygous carriers of the mutation. To the best of our knowledge, this is the first case when a mutation was detected in unrelated, unaffected members of the same community, suggesting the existence of a founder effect in this population. A founder effect was also suspected in the Italian population, where the p.Arg527His LMNA mutation was detected in nine MAD patients from five families originating from sparsely populated area in central Italy, with a probably high percentage of consanguineous marriages. However, this was not confirmed.1
In summary, the main finding reported in this work is a novel c.1580G>T mutation of LMNA causing replacement of arginine 527 by leucine. This mutation was found in three patients of the Egyptian origin, suffering from MADA syndrome and from progeria. We also show that the percentage of mutation carriers in Northeast Egypt might be approximately 1.12%, suggesting the presence of a founder effect. These results deserve a detailed analysis.
References
Novelli G, Muchir A, Sangiuolo F et al: Mandibuloacral dysplasia is caused by a mutation in LMNA encoding lamin A/C. Am J Hum Genet 2002; 71: 426–431.
Agarwal AK, Fryns JP, Auchus RJ, Garg A : Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet 2003; 12: 1995–2001.
Shen JJ, Brown CA, Lupski JR, Potocki L : Mandibuloacral dysplasia caused by homozygosity for the R527H mutation in lamin A/C. J Med Genet 2003; 40: 854–857.
Garavelli L, D'Apice MR, Rivieri F et al: Mandibuloacral dysplasia type A in childhood. Am J Med Genet A 2009; 149A: 2258–2264.
Garg A, Cogulu O, Ozkinay F, Onay H, Agarwal AK : A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia. J Clin Endocrinol Metab 2005; 90: 5259–5264.
Lombardi F, Gullotta F, Columbaro M et al: Compound heterozygosity for mutations in LMNA in a patient with a myopathic and lipodystrophic mandibuloacral dysplasia type A phenotype. J Clin Endocrinol Metab 2007; 92: 4467–4471.
Agarwal AK, Kazachkova I, Ten S, Garg A : Severe mandibuloacral dysplasia-associated lipodystrophy and progeria in a young girl with a novel homozygous Arg527Cys LMNA mutation. J Clin Endocrinol Metab 2008; 93: 4617–4623.
Plasilova M, Chattopadhyay C, Pal P et al: Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome. J Med Genet 2004; 41: 609–614.
Zirn B, Kress W, Grimm T, Berthold LD et al: Association of homozygous LMNA mutation R471C with new phenotype: mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy. Am J Med Genet A 2008; 146A: 1049–1054.
Verstraeten VL, Broers JL, van Steensel MA et al: Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum Mol Genet 2006; 15: 2509–2522.
Cao H, Hegele RA : LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann-Rautenstrauch progeroid syndrome (MIM 264090). Hum Genet 2003; 48: 271–274.
Bonne G, Di Barletta MR, Varnous S et al: Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 1999; 21: 285–288.
Navarro CL, De Sandre-Giovannoli A, Bernard R et al: Lamin A and ZMPSTE24 (FACE-1) defect cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet 2004; 13: 2493–2503.
Ramensky V, Bork P, Sunyaev S : Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002; 30: 3894–3900.
Ferrer-Costa C, Gelpí JL, Zamakola L, Parraga I, de la Cruz X, Orozco M : PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 2005; 21: 3176–3178.
Reumers J, Maurer-Stroh S, Schymkowitz J, Rousseau F : SNPeffect v2.0: a new step in investigating the molecular phenotypic effects of human non-synonymous SNPs. Bioinformatics 2006; 22: 2183–2185.
Ng PC, Henikoff S : Predicting deleterious amino acid substitutions. Genome Res 2001; 11: 863–874.
Cheng J, Randall A, Baldi P : Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 2006; 62: 1125–1132.
Dehouck Y, Kwasigroch JM, Gilis D, Rooman M : PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics 2011; 12: 151.
Parthiban V, Gromiha MM, Schomburg D : CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res 2006; 34: W239–W242.
Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G : Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 2010; 11: 548.
Bao L, Zhou M, Cui Y : nsSNPAnalyzer: identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res 2005; 33: W480–W482.
Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R : Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat 2009; 30: 1237–1244.
Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ : Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev 2006; 86: 967–1008.
Worman HJ, Bonne G : “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res 2007; 313: 2121–2133.
Raffaele Di Barletta M, Ricci E, Galluzzi G et al: Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet 2000; 66: 1407–1412.
Muchir A, Bonne G, van der Kooi AJ et al: Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet 2000; 9: 1453–1459.
Quijano-Roy S, Mbieleu B, Bönnemann CG et al: De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol 2008; 64: 177–186.
Fatkin D, MacRae C, Sasaki T et al: Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999; 341: 1715–1724.
De Sandre-Giovannoli A, Chaouch M, Kozlov S et al: Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 2002; 70: 726–736.
Shackleton S, Lloyd DJ, Jackson SN et al: LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet 2000; 24: 153–156.
De Sandre-Giovannoli A, Bernard R, Cau P et al: Lamin A truncation in Hutchinson-Gilford progeria. Science 2003; 300: 2055.
Eriksson M, Brown WT, Gordon LB et al: Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423: 293–298.
Hennekam RC : Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A 2006; 140: 2603–2624.
Verstraeten VL, Broers JL, Ramaekers FC, van Steensel MA : The nuclear envelope, a key structure in cellular integrity and gene expression. Curr Med Chem 2007; 14: 1231–1248.
Maraldi NM, Capanni C, Cenni V, Fini M, Lattanzi G : Laminopathies and lamin-associated signaling pathways. J Cell Biochem 2011; 112: 979–992.
Scharner J : Gnocchi VF, Ellis JA, Zammit PS: Genotype-phenotype correlations in laminopathies: how does fate translate? Biochem Soc Trans 2010; 38: 257–262.
Cabanillas R, Cadinanos J, Villameytide JAF et al: Nestor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet 2011A; 155: 2617–2625.
Carvalho DR, Pinnola GC, Ferreira DR et al: Mandibular hypoplasia in fibrodysplasia ossificans progressiva causing obstructive sleep apnoea with pulmonary hypertension. Clin Dysmorphol 2010; 19: 69–72.
Dhe-Paganon S, Werner ED, Chi YI, Shoelson SE : Structure of the globular tail of nuclear lamin. J Biol Chem 2002; 277: 17381–17384.
Krimm I, Ostlund C, Gilquin B et al: The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 2002; 10: 811–823.
Ferrè F, Clote P : DiANNA 1.1: an extension of the DiANNA web server for ternary cysteine classification. Nucleic Acids Res 2006; 34: (Web Server issue) W182–W185.
Cheng J, Saigo H, Baldi P : Large-scale prediction of disulphide bridges using kernel methods, two-dimensional recursive neural networks, and weighted graph matching. Proteins 2006; 62: 617–629.
Acknowledgements
This work was in part supported by the statutory funds from the Mossakowski Medical Centre, Polish Academy of Sciences (MPK, AMP). We also acknowledge the support of the 7th Framework Programme of the European Commission Grant HEALTH-PROT, contract number 229676 (LK and JMB).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Al-Haggar, M., Madej-Pilarczyk, A., Kozlowski, L. et al. A novel homozygous p.Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome. Eur J Hum Genet 20, 1134–1140 (2012). https://doi.org/10.1038/ejhg.2012.77
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.77
Keywords
This article is cited by
-
Single-nucleotide polymorphisms in a cohort of significantly obese women without cardiometabolic diseases
International Journal of Obesity (2019)
-
Bicodon bias can determine the role of synonymous SNPs in human diseases
BMC Genomics (2017)
-
Genome-wide association analysis identifies genetic variations in subjects with myalgic encephalomyelitis/chronic fatigue syndrome
Translational Psychiatry (2016)
-
Mandibuloacral dysplasia type A-associated progeria caused by homozygous LMNA mutation in a family from Southern China
BMC Pediatrics (2014)
