
Abstract
The hyper IgM syndromes (HIGM) are a group of primary immune deficiency disorders characterized by defective CD40 signaling by B cells affecting class switch recombination and somatic hypermutation. As a consequence, patients with HIGM have decreased concentrations of serum IgG and IgA and normal or elevated IgM, leading to increased susceptibility to infections. The most common HIGM syndrome is X-linked and due to mutations of CD40 ligand (CD40L) expressed by activated CD4+ T lymphocytes. Four other genes, expressed by B cells, have been associated with the HIGM phenotype. Mutations of CD40, the receptor for CD40L, cause a rare autosomal form of HIGM with a clinical phenotype similar to CD40L deficiency. Mutations of Activation-Induced Cytidine Deaminase (AICDA) and Uracil (DNA) Glycosylase (UNG), both expressed by follicular B lymphocytes, lead to defective class switch recombination and somatic hypermutation. Mutations of Nuclear Factor κB Essential Modulator (NEMO), an X-chromosome associated gene, result in hypohidrotic ectodermal dysplasia and immune deficiency. Thus, the molecular definition of these rare primary immune deficiency disorders has shed light on the complex events leading to the production of high-affinity, antigen-specific antibodies of different isotypes.
Similar content being viewed by others

Main
In 1961, Rosen et al. (1) described two brothers with recurrent infections, and Burtin (2) reported a similar patient who had low levels of 7S gamma-globulin (IgG) and elevated 19S gamma-globulin (IgM). In view of the dissociation between normal or elevated IgM and low-to-undetectable IgG and IgA, this syndrome was originally termed “dysgammaglobulinemia.” A World Health Organization working party, in 1974, named the syndrome immunodeficiency with hyper IgM (HIGM) (3). The nature of the immune defect(s) in HIGM remained elusive. For more than 20 y it was hypothesized that B-lymphocytes from HIGM patients had an intrinsic inability to undergo Ig isotype switching (4). The observation that many patients, especially those with the X-linked form, were also susceptible to opportunistic infections should have pointed to a possible T-cell defect. The important role of defective T helper cells was recognized in 1986, when the observation was made that B cells from male HIGM patients differentiated into IgG secreting cells if co-cultured in vitro with T lymphoblasts from a patient with a Sézary-like syndrome (5). Recently, as more genes were found to be mutated in the syndrome with different clinical presentations, it was suggested to change the name to “defective Ig class switch recombination.”
The gene responsible for the X-linked form of HIGM (XHIGM) was mapped to the long arm of the X-chromosome (Xq26–27) and subsequently identified as CD40ligand (CD40L), expressed by activated CD4+ T cells. CD40L is crucial for T-B cell interaction by its binding to CD40, constitutively expressed by B cells. In 1993, five groups independently and simultaneously showed that mutation in the CD40L gene is the molecular defect responsible for XHIGM (6–10). Subsequent sequence analysis of CD40L revealed that, although most patients with HIGM had mutations in this gene, there was a subgroup of HIGM patients with normal CD40L, including families in which autosomal recessive inheritance was evident. To date, five different genetic defects leading to HIGM have been identified, providing new insight into the complicated process of generating high-affinity antibodies of various Ig isotypes.
Antibody-mediated immune responses play a critical role in the defense against extracellular pathogens and many viruses. The primary antibody repertoire, generated in the bone marrow B cells by means of V(D)J recombination, allows genomic rearrangement between variable (V), diversity (D), and joining (J) gene elements of the Ig heavy and light chain genes. As a result of this process, IgM and IgD antibodies of low avidity are generated, largely in an antigen-independent way. Once IgM-expressing B cells engage antigen, two additional genetic alterations occur to improve specificity and avidity of the antibody to specific microorganisms. This secondary antibody repertoire is generated in the peripheral lymphoid organs and is antigen and T-cell dependent, mainly through the interaction between CD40L (CD154) expressed by activated T cell and CD40 expressed by B cells.
The first modification of the primary IgM antibody response involves CSR. In this process, the constant region of the μ heavy chain is replaced by a downstream heavy chain, e.g. Cγ, Cα, or Cε, leading to the generation of IgG, IgA, or IgE, respectively, with a corresponding change in antibody effector function. This is accomplished through recombination between the switch element upstream of Cγ and that located 5′ to the downstream heavy chain to which rearrangement is targeted (11). The intervening sequences between these two switch regions are deleted.
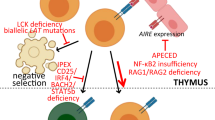
A second antigen-dependent B-cell alteration, SHM, involves the introduction of point mutations at a high rate in the V regions of the Ig genes, resulting in an expansion of the antibody repertoire by allowing the selection of high-affinity antigen-specific antibodies (12). Both CSR and SHM require transcription through target S and V regions on V(D)J exons and DNA editing, which requires two crucial enzymes expressed by germinal center B cells, AID and UNG. Although representing unique modification processes, CSR and SHM are interdependent and nonredundant (13). Defects in any of these events result in antibody deficiency affecting CSR and SHM, collectively referred to as hyper IgM (HIGM) syndromes (14). The characterization of the molecular defects (Table 1, Fig. 1) has been instrumental in unraveling the molecular events leading to antibody diversity and the generation of high-affinity antibodies (15,16).

Schematic representation of the various known molecular defects leading to hyper IgM syndromes.
X-Linked Hyper IgM Due to CD40L Deficiency
The X-linked form of HIGM (XHIGM, HIGM-1) is most common and accounts for about 65–70% of all cases with the HIGM phenotype. Sequence analysis of CD40L and the development of specific disease registries (17–19) have helped in delineating the clinical features of XHIGM. Most, but not all, affected males present in infancy with upper and lower respiratory tract infections. In contrast to most other forms of hypogammaglobulinemia, patients with X-linked HIGM are susceptible to interstitial pneumonia caused by PCP, which can be the presenting symptom (18,19). PCP has been reported in up to 40% of all patients with mutations of CD40L. Diarrhea, present in 50% of the patients, can be acute or chronic and is often associated with cryptosporidium infection that may contribute to the high frequency of sclerosing cholangitis, a severe and often fatal complication. An unusual susceptibility to liver and biliary tract tumors has also been reported (20). Lymph nodes of patients with CD40L deficiency lack germinal centers, attributed to ineffective CD40L/CD40 interaction in the extrafollicular areas, resulting in poor recruitment of germinal center precursors (21).
Autoimmune manifestations are quite common and include thrombocytopenia, seronegative arthritis, and inflammatory bowel disease (18). Neutropenia, which is present in two out of three patients (18) does not seem to be associated with autoantibodies. It can be found in patients who have also chronic parvovirus B19 infection (22). Like all forms of HIGM, patients with mutations of CD40L have markedly reduced levels of serum IgG, IgA, and IgE with normal to elevated IgM and a normal number of circulating B cells, although the proportion of IgD-, CD27+ isotype switched memory B cells is markedly decreased. CD40 signaling is normal as demonstrated by the in vitro production of IgG and IgE when peripheral blood lymphocytes are incubated with antiCD40 or soluble CD40L in the presence of lymphokines (6,7,9). It appears that serum IgM levels increase with age, particularly if initiation of IVIG substitution therapy is delayed (18), indicating that an increase in IgM reflects chronic antigen stimulation rather than a direct effect of the molecular defect.
Whereas the antibody deficiency is considered to be a direct consequence of defective CD40 cross-linking by CD40L, resulting in abnormal CD40 signaling in B cells, the opportunistic infections and increased risk of autoimmune diseases and malignancies are the consequence of the defective T-cell/antigen-presenting cell interaction due to absence of CD40L/CD40 binding. In normal subjects, but not XHIGM patients, activation of T cells is further enhanced by the expression of B7 on the surface of stimulated antigen-presenting cells, a molecule that can deliver a co-stimulatory signal back to T cells via CD28, resulting in optimal T-cell function (23). A direct signal for T cells, via the CD40L intracellular tail, initiated by the engagement of CD40L/CD40 has been postulated by Brenner and coworkers (24) (see below).
The gene responsible for XHIGM, CD40L, was discovered more than 10 y ago (6–10) and belongs to the superfamily of tumor necrosis factor (also known as TNFSFS). The human CD40L protein is 261 amino acid long with a short intracytoplasmic tail (amino acid 1–22), a transmembrane region (amino acid 23–46), and an extracellular domain that shares homology to tumor necrosis factor-α. Crystal structure analysis of the extracellular region shows that both hydrophobic and hydrophilic regions are involved in CD40 binding (25).
The more than 100 unique mutations of CD40L reported (16,26,27; HDO, unpublished data) are scattered throughout the entire gene and may affect the intracellular tail, the trans-membrane region, or, most often, the extracellular domain containing the CD40 binding region. The mutations most frequently observed are those resulting in frameshift and early termination (41%) followed by nonsense mutations (26%) and missense mutations (20%). In a few families, mutations involving splice sites may result in the generation of small amounts of wild-type CD40L, associated with a milder clinical phenotype (26).
The clinical diagnosis has to be confirmed by demonstrating a defect in the expression of CD40L by activated peripheral blood CD4+ lymphocytes using anti-CD40L MAb or a soluble CD40-Ig fusion protein and flow cytometry. However, because most mutations result in nonfunctional/truncated protein expression, activated CD4+ lymphocytes from approximately 20% of XHIGM patients can bind anti-CD40L MAb and therefore have to be identified by binding CD40-Ig fusion protein or by sequence analysis. In some instances, when small amounts of wild-type CD40L are expressed by activated CD4+ cells, the CD40-Ig fusion protein may be bound, although at low intensity. Therefore, the final molecular diagnosis depends on sequence analysis using cDNA or genomic DNA (26).
There is no strong genotype/phenotype correlation in XHIGM, however, several mutations have been reported to be associated with a mild phenotype, late onset, and prolonged survival (21).
Conservative treatment consists of prophylactic IVIG at doses of 400–600 mg/kg per month. Infants with XHIGM are particularly susceptible to PCP and should receive prophylaxis with trimethoprim-sulfamethoxazole. Patients with persistent severe neutropenia are candidates for treatment with granulocyte colony-stimulating factor (G-CSF) (28). Prevention of liver disease is of prime concern and necessitates careful monitoring of liver and biliary tract function, which may require ultrasound examinations and biopsies. Because cryptosporidium infection has been associated with chronic cholangitis, avoiding this parasite is important, e.g. use of boiled or filtered water is recommended.
Nevertheless, the long-term prognosis of XHIGM is guarded. A multicenter European study suggests that only 20% of the patients survive beyond 25 y of age (17). The causes of death included infections during early life, liver disease, and malignancies. For these reasons, stem cell transplantation has been advocated (30), using HLA-identical siblings, matched unrelated donors, or partially matched umbilical cord blood. Although preliminary reports were encouraging (29,30), a recent study of 38 patients with XHIGM who underwent BMT in Europe showed only 68% survival rate (31). If the patient is relatively young (≤8 y of age) and without serious infection and if an optimal donor (e.g. matched sibling) is available, stem cell transplantation should be seriously considered. Gene therapy will be more complicated than that of X-linked severe combined immunodeficiency. Because the expression of the CD40L gene is highly regulated, a dominant negative situation may occur and overexpression may be detrimental (32).
Autosomal Recessive HIGM Due to CD40 Deficiency
This rare form of HIGM (HIGM-3), recognized to-date in only four patients from three unrelated families (33,34), with autosomal recessive mode of inheritance is due to mutations in CD40, a member of the tumor necrosis factor receptor superfamily. CD40 is expressed constitutively on the surface of B-lymphocytes, mononuclear phagocytes, dendritic cells, and activated epithelial cells (33). Activated T cells that express CD40L will engage and cross-link CD40 on resting B cells and provide key signals for B cells to generate a set of proteins/enzymes required for CSR and SHM (35).
Similar to CD40L deficiency, patients with CD40 mutations present during infancy with severe clinical symptoms, including opportunistic infections and failure to thrive, suggestive of combined immunodeficiency. The lack of activation of monocytes and dendritic cells in the absence of CD40 may account for opportunistic infections in both CD40L and CD40 deficiencies. Cross-linking of CD40 is required for the initiation of CD40 signaling, which results in increased expression of B7 by B cells (23,35). The interaction of B7 with the T-cell surface molecules CD28 and CTLA-4 results in additional T-cell activation. Finally, although the signaling within the T cell mediated by CD40L is still controversial, there is experimental evidence that co-stimulation of T cells via CD40L following CD40L–CD40 interaction is required to initiate direct activation of T lymphocytes by tyrosine phosphorylation of cellular proteins including PLCγ; however, no binding partner for CD40L has been identified in CD4+ T cells (24).
The CD40-deficient patients identified to date were found to have a complete lack of CD40 expression on the surface of B lymphocytes and monocytes. B-cell stimulation in vitro with anti CD40 and IL-10 failed to induce IgA or IgG production (33) in contrast to B cells from XHIGM patients who do (6,7,9). Similar to XHIGM patients, CD40 deficient infants have decreased numbers of IgD−, CD27+ memory B cells.
Treatments include IVIG infusions every 3–4 wk, PCP prophylaxis, and attention to nutrition. Stem cell transplantation is expected to be less successful (36), since it restores CD40 expression only for hematopoietic stem cell–derived cell lineages and not for other CD40-expressing cell types.
Autosomal-Recessive Hyper-IgM Syndrome Due to AID Deficiency
Following the discovery of the molecular basis of XHIGM, a number of investigators identified male and female patients with HIGM, normal CD40L, increased susceptibility to bacterial, but not opportunistic infections, and in some families an autosomal-recessive pattern of inheritance (37–39). In 2000, Revy et al. (40) studied such a group of HIGM patients and found mutations within Activation-Induced Cytidine Deaminase (AICDA), the gene encoding AID.
AID-deficient (HIGM-2) patients present during early childhood with recurrent bacterial sinorespiratory and gastrointestinal tract infections. However, because of the milder disease and lack of opportunistic infections, many patients with mutations of AID are not recognized as having immunodeficiency until the second or third decade of life (41) (Table 1). Similar to patients with CD40L mutations, AID-deficient patients present with markedly diminished levels of IgG and IgA, and with normal or elevated IgM. Specific antibodies of the IgG class to T-cell-dependent protein antigens are absent, whereas IgM isohemagglutinins are present. The numbers of CD19+ B cells and CD27+ memory B cells are normal, and T-cell immunity is universally intact (40). A characteristic clinical finding, present in half of the patients with AID deficiency, is lymphoid hyperplasia due to the presence of giant germinal centers filled with proliferating B-lymphocytes co-expressing IgM, IgD, and CD38 (42).
The AICDA gene was cloned by subtractive hybridization between murine lymphoma B cells that had or had not been induced to undergo CSR in vitro. AID expression is restricted to LPS-activated B cells undergoing in vitro Ig CSR and to germinal center B cells (43). The gene encoding human AID is located on chromosome 12p13, includes 5 exons, and generates a 198 amino acid protein. Mutations of AID, most often homozygous, rarely compound heterozygous, have been found throughout the gene, predominantly in exon 3. They include amino acid substitutions, premature stop codons, and deletions (40,41). AID contains an active site for cytidine deamination that is conserved in members of the large cytidine deaminase family.
Originally, AID was considered to be an RNA-editing enzyme involved in one or several target mRNA (42–44). However, recent data have provided strong evidence that AID acts directly on DNA (45). In this model, AID is expected to convert deoxycytidine (dC) into deoxyuridine (dU) in one strand of DNA (46). The strongest evidence that AID acts directly on DNA has been provided by the recent description of a partial impairment in CSR associated with a normal frequency but skewed pattern of SHM in mice deficient of uracil N-glycosylase (UNG) (47) and of a new HIGM condition characterized by a profound defect in CSR at a DNA precleavage step and a partial disturbance of the SHM pattern caused by mutations of the human UNG gene (48) (see below). Deamination of cytidine represents a physiologic trigger for base excision repair, which is a crucial step in DNA cleavage. Thus, a single protein, AID, differentially regulates V-region point mutation and S-region recombination. It was recently reported that AID requires interaction with specific co-factor(s) to induce CSR (49). It was also shown that the block of CSR caused by AID deficiency occurs before the generation of DNA double-strand breaks in the switch μ-region (50). Thus, although the precise mechanism by which AID exerts its function is still not completely understood, its essential role in CSR and SHM has been demonstrated convincingly. A positive in vitro class switching with anti-CD40 and cytokines essentially rules out the diagnosis of AID deficiency
The diagnosis of AID deficiency should be considered in patients with abnormal serum Ig levels, suggesting the HIGM phenotype in the presence of normal CD40L expression and/or sequence and the inability of peripheral blood lymphocytes to generate in vitro Ig other than IgM if stimulated with antiCD40 and lymphokines. To confirm the molecular diagnosis, it is necessary to demonstrate mutations within the AID gene.
Treatment with regular infusions of IVIG (400–600 mg/kg/mo) is effective in reducing infections but does not affect lymphoid hyperplasia.
Autosomal Recessive HIGM Due to UNG Deficiency
In a collaborative study to search for the molecular defect in a group of HIGM patients with normal CD40L, CD40, and AID, three patients, one each from France, Japan, and the United States, were found to have mutations affecting UNG (48). The clinical phenotype of UNG deficiency resembled patients with AID deficiency, including susceptibility to bacterial infections, lymphoid hyperplasia, increased serum IgM concentrations, and profoundly decreased serum IgG and IgA levels. Numbers of B- and T-cell subsets were normal. Memory B cells (CD 27+) were present in normal numbers in two of the three UNG-deficient patients. Their B cells were unable to undergo CSR in vitro following CD40 activation with soluble CD40L and lymphokines. RNA transcripts of the AICDA gene were present, indicating that the block in CSR was not a consequence of a defect in the signaling events leading to AID expression. However, after activation via CD40, B cells from UNG-deficient patients failed to generate double-stranded DNA breaks in S-regions, similar to patients with AID deficiency. When SHM was analyzed in the VH region of IgM in purified memory B cells, a SHM pattern was found in which mutations at dG and dC residues were biased toward transition, whereas at dA and dT residues, the ratio of transitions to transversions was similar to control values (48). Similar immunologic abnormalities as well as partially defective CSR and a bias pattern of SHM toward transitions at dG-dC nucleotides were reported in UNG-deficient mice (47). As in mice, human UNG has two promoters and two alternative splice products: the mitochondrial isoform, which is ubiquitously expressed, and the nuclear isoform, which is strongly expressed in proliferating cells (51,52). In each of the three patients, mutations were located in the catalytic domain of UNG. One patient was a compound heterozygote for a C deletion in one allele and a TA deletion in the other. The second patient was homozygous for an amino acid substitution (F251S). The third patient was homozygous for an AT deletion in exon 2. UNG, expressed only in activated CD19+ B cells but not in T cells, is also present in EBV-transformed normal B-lymphoblastoid cell lines but was found consistently absent in EBV-transformed B-lymphoblastoid cell lines derived from the three UNG-deficient patients (48).
Although the exact mechanism by which UNG exerts its function is not completely understood (53), analysis of UNG-deficient mice and humans supports the model of CSR in which AID directly deaminases cytidine into uracil residues in the active S-regions, followed by uracil removal mediated by UNG, leading to an abasic site that is essential for SHM as well as for switch recombination.
HIGM with Defective CSR but Normal SHM
Molecular analysis of large cohorts of patients with the HIGM phenotype suggested that 20–30% have normal CD40L, CD40, AID, and UNG genes. Recently, a subset of these molecularly undefined patients has been identified as having in common a defect of CSR while SHM was intact, accounting for approximately 10% of HIGM patients (54). Most of the cases were sporadic with no family history of immunodeficiency, but in a few an autosomal recessive mode of inheritance was suggested.
Similar to patients with defective AID or UNG, this subgroup of HIGM patients suffers from recurrent bacterial infections, beginning during childhood without particular susceptibility to opportunistic infections (Table 1), suggesting that T-cell immunity is unaffected. In accordance with a clinical course described as “slightly milder” than that of AID deficiency, the mean age of diagnosis was relatively late at 8.8 y. Like subjects with AID and UNG deficiency, this subgroup of HIGM patients often develops lymphoid hyperplasia but without the giant germinal centers typical of AID-deficient patients. The CSR defect is less severe, as suggested by the occasional presence of residual IgG. Numbers of B and T cells, including subsets and T-cell function, are normal. Of note, more than 60% of this cohort of HIGM patients have low numbers of memory B cells (54).
Functional in vitro studies revealed that B-cell CSR was intrinsically impaired, but SHM assessment in the variable region of the Ig heavy chain gene was completely normal. In vitro studies suggested that the molecular defect responsible for this group of patients occurs downstream of AID, as the AID gene was expressed normally and AID-induced DNA double-strand breaks in the switch (S) regions of the Ig heavy chain locus, a requirement for successful CSR, were normally detected. Although this group of CSR-deficient patients has a characteristic phenotype and normal AID and UNG, it is unlikely that they all will have a single gene defect. It remains to be seen whether at least a subset of this group will turn out to have a unique molecular defect involving one or more gene(s) that are directly involved in CSR or in CSR-specific DNA repair, or if these patients have a multifactorial disorder involving multiple predisposition genes.
X-Linked HIGM Due to Mutations of NEMO
Hypohidrotic (anhidrotic) EDA is a rare syndrome associated with abnormal development of hair, teeth, and sweat glands (55). Most patients with EDA are immunologically normal and have mutations caused by the ectodysplasin-A gene, EDA1, located on the X chromosome (56), or mutations of the ectodysplasin-A receptor (EDAR) an autosomal recessive disorder (57). A subset of the X-linked form of EDA has been found to be associated with immunodeficiency (EDA-ID), characterized by susceptibility to bacterial infections, including atypical mycobacteria (58–60). Many, but not all, of the EDA-ID patients have dysgammaglobulinemia characterized by decreased IgG and decreased or elevated IgM and IgA. The gene responsible for this X-linked form of HIGM has been identified as the NF κB essential modulator (NEMO), also designated as IKK-γ (61–63). Mutations of NEMO interfere with NF-κB activation (64). In resting cells, NF-κB activity is inhibited by one of several inhibitors (IκB). The cytoplasmic association of IκB and NF-κB is terminated by the phosphorylation of IκB, which leads to its cytoplasmic degradation. The release of the NF-κB complex from IκB allows NF-κB to translocate to the nucleus and activate transcription. Phosphorylation of IκB is mediated by IκB kinase (IKK), a complex consisting of IKKα, IKKβ, and IKKγ (the latter also known as NEMO). A variety of cell surface receptors have been identified that can induce signaling pathways that result in the activation of the IKK complex leading to the phosphorylation of IκB. The cell surface receptors involved in this process include TNF superfamily receptors such as EDAR, toll-like receptors, and CD40. Mutation of NEMO is therefore of crucial importance for the clinical phenotypes observed in EDA-ID. For instance, following cell activation by cross-linking CD40, the IKK signalosome is activated, leading to the phosphorylation and degradation of the NF-κB inhibitory protein, IκB, and thus to the translocation of NF-κB to the nucleus. If NF-κB translocation is defective, multiple NF-κB-dependent proteins, including AID and UNG, cannot be expressed.
The NEMO gene consists of 10 exons and encodes a protein (IKK-γ) that binds IKK-α and IKK-β, and thus is responsible for maintaining a functional IKK complex. The majority of patients with EDA-ID were found to have a point mutation in the C-terminal portion of the NEMO gene that encodes a zinc finger domain believed to be an important modulator for upstream activators (61–63). These hypomorphic NEMO mutations are associated with EDA-ID phenotype and sometimes, in addition, with osteopetrosis and lymphedema (OL-EDA-ID). In contrast, mutations of NEMO that result in “loss of function” are the cause of incontinentia pigmenti in carrier females and lead to embryonic death in males (65). Recently, an autosomal dominant form of EDA-ID has been reported, caused by the mutation of one allele of IκBα, a subunit of the IκB complex (66). Thus, defective NFκB translocation from the cytoplasm to the nucleus can be caused by mutation of more than one gene, and not only interferes with T- and B-cell activation, including CD40 mediated CSR and SHM, but may affect multiple ectoderm-derived structures.
Abbreviations
- AICDA:
-
(AID), activation-induced cytidine deaminase
- BMT:
-
bone marrow transplantation
- CD40L:
-
CD40 ligand
- CSR:
-
class switch recombination
- EDA:
-
ectodermal dysplasia
- EDA-ID:
-
ectodermal dysplasia with immunodeficiency
- HIGM:
-
hyper IgM syndrome
- IKK:
-
Ikappa B kinase
- IVIG:
-
intravenous Ig
- NEMO:
-
nuclear factor κB essential modulator
- PCP:
-
Pneumocystis carinii pneumonia
- SHM:
-
somatic hypermutation
- UNG:
-
uracil DNA glycosylase
- XHIGM:
-
X-linked hyper IgM syndrome
References
Rosen FS, Kevy SV, Merler E, Janeway CA, Gitlin D 1961 Recurrent bacterial infections and dysgamma-globulinemia: deficiency of 7S gamma-globulins in the presence of elevated 19S gamma-globulins. Report of two cases. Pediatrics 28: 182–195
Burtin P 1961 [An example of atypical agammaglobulinemia (a case of severe hypogammaglobulinemia with increase of the beta-2 macroglobulin).]. Rev Franc Etud Clin Biol 6: 286–289
Cooper MD, Faulk WP, Fudenberg HH, Good RA, Hitzig W, Kunkel HG, Roitt IM, Rosen FS, Seligmann M, Soothill JF 1974 Meeting report of the second international workshop on primary immunodeficiency disease in man held in St. Petersburg, Florida, February, 1973. Clin Immunol Immunopathol S: 416–445
Notarangelo LD, Duse M, Ugazio AG 1992 Immunodeficiency with hyper IgM (HIM). Immunodefic Rev 3: 101–121
Mayer L, Kwan SP, Thompson C, Ko HS, Chiorazzi N, Waldmann T, Rosen F 1986 Evidence for a defect in “switch” T cells in patients with immunodeficiency and hyperimmunoglobulinemia M. N Engl J Med 314: 409–413
Allen RC, Armitage RJ, Conley ME, Rosenblatt H, Jenkins NA, Copeland NG, Bedell MA, Edelhoff S, Disteche CM, Simoneaux DK, Fanslow WC, Belmont J, Spriggs MK 1993 CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science 259: 990–993
Aruffo A, Farrington M, Hollenbough D, Li X, Milatovich A, Nonoyama S, Bajorath J, Grosmaire LS, Stenkamp R, Neubauer M, Roberts RL, Noelle RJ, Ledbetter JA, Francke U, Ochs HD 1993 The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell 72: 291–300
DiSanto JP, Bonnefoy JY, Gauchat JF, Fischer A, De Saint Basile G 1993 CD40 ligand mutations in x-linked immunodeficiency with hyper-IgM. Nature 361: 541–543
Fuleihan R, Ramesh N, Loh R, Jabara H, Rosen RS, Chatila T, Fu SM, Stamenkovic I, Geha RS 1993 Defective expression of the CD40 ligand in X chromosome-linked immunoglobulin deficiency with normal or elevated IgM. Proc Natl Acad Sci U S A 90: 2170–2173
Korthäuer U, Graf D, Mages HW, Brière F, Padayachee M, Malcolm S, Ugazio AG, Notarangelo LD, Levinsky RJ, Kroczek RA 1993 Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature 361: 539–541
Kenter AL 2003 Class switch recombination: after the dawn of AID. Curr Opin Immunol 15: 190–198
Papavasiliou FN, Schatz DG 2002 Somatic hypermutation of immunoglobulin genes; merging mechanisms for genetic diversity. Cell 109: S35–S44
Kinoshita K, Honjo T 2001 Linking class-switch recombination with somatic hypermutation. Nat Rev Mol Cell Biol 2: 493–503
de Villartay JP, Fischer A, Durandy A 2003 The mechanisms of immune diversification and their disorders. Nat Rev Immunol 3: 962–972
Manis JP, Alt FW 2003 Novel antibody switching defects in human patients. J Clin Invest 112: 19–22
Gulino AV, Notarangelo LD 2003 Hyper IgM syndromes. Curr Opin Rheumatol 15: 422–509
Notarangelo LD, Peitsch MC 1996 CD40lbase: a database of CD40L gene mutations causing X-linked hyper-IgM syndrome. Immunol Today 17: 511–516
Levy J, Espanol-Boren T, Thomas C, Fischer A, Tovo P, Bordigoni P, Resnick I, Fasth A, Baer M, Gomez L, Sanders EA, Tabone MD, Plantaz D, Etzioni A, Monafo V, Abinun M, Hammarstrom L, Abrabamsen T, Jones A, Finn A, Klemola T, DeVries E, Sanal O, Peitsch MC, Notarangelo LD 1997 Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr 131: 47–54
Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, Stiehm ER, Conley ME 2003 The X-linked hyper IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore) 82: 373–384
Hayward AR, Levy J, Facchetti F, Notarangelo L, Ochs HD, Etzioni A, Bonnefoy JY, Cosyns M, Weinberg A 1997 Cholangiopathy and tumors of the pancreas, liver, and biliary tree in boys with X-linked immunodeficiency with hyper-IgM. J Immunol 158: 977–983
Facchetti F, Appianai C, Salvi L, Levy J, Notarangelo LD 1995 Immunohistologic analysis of ineffective CD40-CD40 ligand interaction in lymphoid tissues from patients with X-linked immunodeficiency with hyper-IgM. Abortive germinal center cell reaction and severe depletion of follicular dendritic cells. J Immunol 154: 6624–6633
Seyama K, Kobayashi R, Hasle H, Apter AJ, Rutledge JC, Rosen D, Ochs HD 1998 Parvovirus B19-induced anemia as the presenting manifestation of X-linked hyper-IgM syndrome. J Infect Dis 178: 318–324
Grewal IS, Flavell RA 1996 The role of CD40 ligand in costimulation and T-cell activation. Immunol Rev 153: 85–106
Brenner B, Koppenhoefer U, Lepple-Wienhues A, Grassmé H, Müller C, Speer CP, Lang F, Gulbins E 1997 The CD40 ligand directly activates T-lymphocytes via tyrosine phosphorylation dependent PKC activation. Biochem Biophys Res Commun 239: 11–17
Karpusas M, Hsu YM, Wang JH, Thompson J, Lederman S, Chess L, Thomas D 1995 2å crystal structure of an extracellular fragment of human CD40ligand. Structure 3: 1031–1039
Seyama K, Nonoyama S, Gangsaas I, Hollenbaugh D, Pabst HF, Aruffo A, Ochs HD 1998 Mutations of the CD40 ligand gene and its effect on CD40 ligand expression in patients with X-linked hyper IgM syndrome. Blood 92: 2421–2434
Vihinen M, Arredondo-Vega FX, Casanova JL, Etzioni A, Giliani S, Hammarstrom L, Hershfield MS, Heyworth PG, Hsu AP, Lahdesmaki A, Lappalainen I, Notarangelo LD, Puck JM, Reith W, Roos D, Schumacher RF, Schwarz K, Vezzoni P, Villa A, Valiaho J, Smith CI 2001 Primary immunodeficiency mutation databases. Adv Genet 43: 103–188
Wang WC, Cordoba J, Infante AJ, Conley ME 1994 Successful treatment of neutropenia in the hyper-immunoglobulin M syndrome with granulocyte colony-stimulating factor. Am J Pediatr Hematol Oncol 16: 160–163
Hadzic N, Pagliuca A, Rela M, Portmann B, Jones A, Veys P, Heaton ND, Mufti GJ, Mieli-Vergani G 2000 Correction of the hyper-IgM syndrome after liver and bone marrow transplantation. N Engl J Med 342: 320–324
Duplantier JE, Seyama K, Day NK, Hitchcock R, Nelson RP Jr Ochs HD, Haraguchi S, Klemperer MR, Good RA 2001 Immunologic reconstitution following bone marrow transplantation for X-linked hyper IgM syndrome. Clin Immunol 98: 313–318
Gennery AR, Khawaja K, Veys P, Bredius RG, Notarangelo LD, Mazzolari E, Fischer A, Landais P, Cavazzana-Calvo M, Friedrich W, Fasth A, Wulffraat NM, Matthes-Martin S, Bensoussan D, Bordigoni P, Lange A, Pagliuca A, Andolina M, Cant AJ, Davies EG 2004 Treatment of CD40ligand deficiency by haemopoietic stem cell transplantation: a survey of the European experience, 1993–2002. Blood 103: 1152–1157
Seyama K, Osborne WR, Ochs HD 1999 CD40L ligand mutants responsible for X-linked hyper-IgM syndrome associate with wild type CD40 ligand. J Biol Chem 274: 11310–11320
Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, Avanzini MA, Marconi M, Badolato R, Ugazio AG, Levy Y, Catalan N, Durandy A, Tbakhi A, Notarangelo LD, Plebani A 2001 Mutations of CD40 gene causes an autosomal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad Sci U S A 98: 12614–12619
Kutukculer N, Moratto D, Aydinok Y, Lougaris V, Aksoylar S, Plebani A, Genel F, Notarangelo LD 2003 Disseminated cryptosporidium infection in an infant with hyper-IgM syndrome caused by CD40 deficiency. J Pediatr 142: 194–196
Van Essen D, Kikutani H, Gray D 1995 CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature 378: 620–623
Kutukculer N, Aksoylar S, Kensoy S, Cetingul N, Notarangelo LD 2003 Outcome of hematopoietic stem cell transplantation in hyper-IgM syndrome caused by CD40 deficiency. J Pediatr 143: 141–142
Callard RE, Smith SH, Herbert J, Morgan G, Padayachee M, Lederman S, Chess L, Kroczek RA, Fanslow WC, Armitage RJ 1994 CD40 ligand (CD40L) expression and B cell function in agammaglobulinemia with normal or elevated levels of IgM (HIM). Comparison of X-linked, autosomal recessive, and non-X-linked forms of the disease, and obligate carriers. J Immunol 153: 3295–3306
Conley ME, Larche M, Bonagura VR, Lawton AR III Buckley RH, Fu SM, Coustan-Smith E, Herrod HG, Campana D 1994 Hyper IgM syndrome associated with defective CD40-mediated B cell activation. J Clin Invest 94: 1404–1409
Durandy A, Hivroz C, Mazerolles F, Schiff C, Bernard F, Jouanguy E, Revy P, DiSanto JP, Gauchat JF, Bonnefoy JY, Casanova JL, Fischer A 1997 Abnormal CD40 mediated activation pathway in B lymphocytes from patients with hyper-IgM syndrome and normal CD40 ligand expression. J Immunol 158: 2576–2584
Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A 2000 Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper- IgM syndrome (HIGM2). Cell 102: 565–575
Minegishi Y, Lavoie A, Cunningham-Rundles C, Bedard PM, Hebert J, Côté L, Dan K, Sedlak D, Buckley RH, Fischer A, Durandy A, Conley ME 2000 Mutations in activation induced cytidine deaminase in patients with hyper IgM syndrome. Clin Immunol 97: 203–210
Durandy A, Honjo T 2001 Human genetic defects in class-switch recombination (hyper- IgM syndromes). Curr Opin Immunol 13: 543–548
Muto T, Muramatsu M, Taniwaki M, Kinoshita K, Honjo T 2000 Isolation, tissue distribution, and chromosomal localization of the human activation-induced cytidine deaminase (AID) gene. Genomics 68: 85–88
Durandy A 2003 Activation-induced cytidine deaminase: a dual role in class-switch recombination and somatic hypermutation. Eur J Immunol 33: 2069–2073
Petersen-Mahrt SK, Harris RS, Neuberger MS 2002 AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 418: 99–103
Reynaud CA, Aoufouchi S, Faili A, Weill JC 2003 What role for AID: mutator, or assembler of the immunoglobulin mutasome?. Nat Immunol 4: 631–638
Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS 2002 Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol 12: 1748–1755
Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, Ochs HD, Fischer A, Durandy A 2003 Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol 4: 1023–1028
Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, Nonoyama S, Tashiro J, Ikegawa M, Ito S, Kinoshita K, Muramatsu M, Honjo T 2003 AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol 4: 843–848
Catalan N, Selz F, Imai K, Revy P, Fischer A, Durandy A 2003 The block in immunoglobulin class switch recombination caused by activation-induced cytidine deaminase deficiency occurs prior to the generation of DNA double strand breaks in switch mu region. J Immunol 171: 2504–2509
Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan HE 1997 Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res 25: 750–755
Otterlei M, Haug T, Nagelhus TA, Slupphaug G, Lindmo T, Krokan HE 1998 Nuclear and mitochondrial splice forms of human uracil-DNA glycosylase contain a complex nuclear localisation signal and a strong classical mitochondrial localisation signal, respectively. Nucleic Acids Res 26: 4611–4617
Storb U, Stavnezer J 2002 Immunoglobulin genes: generating diversity with AID and UNG. Curr Biol 12: R725–R727
Imai K, Catalan N, Plebani A, Marodi L, Sanal O, Kumaki S, Nagendran V, Wood P, Glastre C, Sarrot-Reynauld F, Hemine O, Forveille M, Revy P, Fischer A, Dunrandy A 2003 Hyper-IgM syndrome type 4 with a B lymphocyte-intrinsic selective deficiency in Ig class-switch recombination. J Clin Invest 112: 136–142
Clarke A, Phillips DI, Brown R, Harper PS 1987 Clinical aspects of X-linked hypohidrotic ectodermal dysplasia. Arch Dis Child 62: 989–996
Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, Ferguson B, Munoz F, Morgan D, Clarke A, Baybayan P, Chen EY, Ezer S, Saarialho-Kere U, de la Chapelle A, Schlessinger D 1996 X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 13: 409–416
Monreal AW, Ferguson BM, Headon DJ, Street SL, Overbeek PA, Zonana J 1999 Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet 22: 366–369
Abinun M, Spickett G, Appleton AL, Flood T, Cant AJ 1996 Anhidrotic ectodermal dysplasia associated with specific antibody deficiency. Eur J Pediatr 155: 146–147
Schweizer P, Kalhoff H, Horneff G, Wahn V, Diekmann L 1999 Polysaccharide specific humoral immunodeficiency in ectodermal dysplasia: case report of a boy with two affected brothers. Klin Pädiatr 211: 459–461
Carrol ED, Gennery AR, Flood TJ, Spickett GP, Abinun M 2003 Anhidrotic ectodermal dysplasia and immunodeficiency: the role of NEMO. Arch Dis Child 88: 340–341
Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, Shapira SK, Farndon PA, Wara DW, Emmal SA, Ferguson BM 2000 A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet 67: 1555–1562
Döffinger R, Smahi A, Bessia C, Geissman F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S, Wood P, Rabia SH, Headon DJ, Overbeek PA, Le Deist F, Holland SM, Belani K, Kumararatne DS, Fischer A, Shapiro R, Conley ME, Reimund E, Kalhoff H, Abinun M, Munnich A, Israel A, Courtois G, Casanova JL 2001 X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB signaling. Nat Genet 27: 277–285
Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W 2001 Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol 2: 223–228
Orange JS, Geha RS 2003 Finding NEMO: genetic disorders of NF-κB activation. J Clin Invest 112: 983–985
Aradhya S, Woffendin H, Jakins T, Bardaro T, Esposito T, Smahi A, Shaw C, Levy M, Munnich A, D'Urso M, Lewis RA, Kenwrick S, Nelson DL 2001 A recurrent deletion in the ubiquitously expressed NEMO (IKK-γ) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum Mol Genet 10: 2171–2179
Courtois G, Smahi A, Reichenbach J, Döffinger R, Cancrini C, Bonnet M, Puel A, Chable-Bessia C, Yamaoka S, Feinberg J, Dupuis-Girod S, Bodemer C, Livadiotti S, Novelli F, Rossi P, Fischer A, Israël A, Munnich A, Le Deist F, Casanova JL 2003 A hypermorphic IκBα mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest 112: 1108–1115
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Etzioni, A., Ochs, H. The Hyper IgM Syndrome—An Evolving Story. Pediatr Res 56, 519–525 (2004). https://doi.org/10.1203/01.PDR.0000139318.65842.4A
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000139318.65842.4A
This article is cited by
-
A Retrospective Study of Clinical and Immunological Features of a Pediatric Population with Talaromyces marneffei Infection
Mycopathologia (2023)
-
Dysbiosis and primary B-cell immunodeficiencies: current knowledge and future perspective
Immunologic Research (2023)
-
X-linked hyper IgM syndrome with severe eosinophilia: a case report and review of the literature
BMC Pediatrics (2022)
-
Autoimmunity in Primary Immunodeficiencies (PID)
Clinical Reviews in Allergy & Immunology (2022)
-
Somatic hypermutation defects in two adult hyper immunoglobulin M patients
Immunologic Research (2022)
