
Abstract
Intravascular lymphoma (IVL) is a rare neoplastic disease characterized by the presence of large malignant lymphoid cells in small vessels. It is often diagnosed at autopsy. Clinical manifestations are typically neurologic and dermatologic. Karyotypic abnormalities have been described in a small number of cases and have revealed complex alterations in the majority of cases. We have identified three cases of IVL with varied clinicopathological findings. Karyotypic analysis was undertaken by standard G-banding and supplemented by multi-colored karyotyping (M-FISH) to decipher the chromosomal content of marker chromosomes and undefined additions. M-FISH clarified the chromosomal abnormalities in two cases and unveiled cryptic translocations der(10)t(10;22), der(17)t(17;22), and balanced t(11;14). Comparison with previously published karyotypes revealed prominent involvement of chromosomes 1, 3, 6, 11, 14, and 18, similar to the pattern of clonal evolution in other B-cell lymphomas. The most frequent alterations seen were −6 or 6q− and +18 or dup(18q), with a minimally deleted region located at 6q21-q23 and a commonly amplified region located at 18q13-q23, respectively. Few differences between the classical and Asian variant of this disease were apparent at the karyotypic level. Cytogenetic analysis of additional cases supplemented by multicolor karyotyping may help identify the full spectrum of genetic alterations associated with IVL and assist in the delineation of the critical mutations associated with initiation and progression of this disease.
Similar content being viewed by others


INTRODUCTION
Intravascular lymphoma (IVL) is a rare disease characterized by the presence of large malignant lymphoid cells in small vessels (1, 2, 3, 4). The malignant cells are usually confined within the vascular lumen (5, 6), and a perivascular infiltration is occasionally seen (7, 8, 9). In about 90% of cases the neoplastic cells are of B-cell origin, whereas rare cases of T-cell IVL have been reported (10, 11). The clinical manifestations are predominantly neurologic and dermatologic (12, 13, 14). A wide range of other organ involvement has been described: adrenal glands, lungs, gastrointestinal system, kidneys, genitourinary tract, and eye (15, 16, 17, 18, 19, 20). At autopsy, involvement of the majority of organs is seen despite the absence of prior clinical manifestations (21, 22). The involvement of hematopoietic tissues (bone marrow, lymph nodes, spleen, and liver) is infrequent (12, 23), and the presence of circulating malignant cells is exceptional (24).
A few cases of IVL associated with hemophagocytic syndrome (HPS) have been described in the literature (25). These cases are predominantly reported in Asian patients. Absence of skin involvement, rarity of neurological symptoms, frequent bone marrow involvement, and constant hepatosplenomegaly characterize these cases (26). It has recently been suggested that cases of IVL associated with HPS could constitute a distinct variant of IVL (27).
Cytogenetic abnormalities in non-Hodgkin’s lymphoma (NHL) have been correlated with histological subtype, immunophenotype, and clinical behavior, and have contributed to the identification of the molecular pathways implicated in lymphoma pathogenesis. Karyotypic data have been described only in a very few cases of IVL (26, 28, 29, 30, 31, 32, 33). All cases have shown an abnormal karyotype with complex chromosomal abnormalities. Highly complex karyotypes are difficult to interpret using conventional G- or R-banding techniques. New techniques such as spectral karyotyping (SKY) and multicolor fluorescence in situ hybridization (M-FISH), which identify each of the 24 human chromosomes in a different color simultaneously, have been useful in analyzing such complex karyotypes. These multicolor karyotyping techniques help to clarify the content of marker chromosomes and to detect subtle chromosomal abnormalities not apparent by conventional karyotyping (34, 35, 36, 37, 38).
Here, we report the clinicopathological and cytogenetic findings using G-banding and M-FISH techniques of three cases of IVL and a review of the previously published karyotype data.
CASE REPORTS
All clinical and laboratory findings of the study cases are summarized in Table 1.
Case 1
A 63-year-old Caucasian woman presented with a 2-month history of weight loss, high-grade fever, skin eruptions, and sudden onset of a paralysis of the lower limbs. On physical examination, she had complete flaccid paralysis of both legs, and deep tendon reflexes were absent in both ankles. Multiple erythematous annular plaques were seen on her back. No hepatomegaly, splenomegaly, or adenopathy was noticed. Complete blood cell count showed white blood cell count of 12 × 109 per liter, Hb of 97 g/L, platelets of 239 × 109 per liter, and leukoerythroblastic reaction. A few atypical lymphocytes were seen on the blood smears. The LDH was elevated at 3990 U/L (normal, <210). Whole-body CT scan and MRI of the lumbar spine did not show any abnormalities. Punch skin biopsy revealed intravascular lymphoid infiltrates. Bone marrow biopsy and immunostain showed atypical B-lymphoid cells with an intravascular pattern. The patient was treated with polychemotherapy according to the ACOP12 protocol. She received six biweekly cycles of chemotherapy. After initial improvement, the patient experienced relapse and died 1 month after the last treatment cycle.
Case 2
A 62-year-old Caucasian man was referred for management of pancytopenia and circulating abnormal cells. He had a 3-month history of anorexia, increasing fatigue, and mild shortness of breath. At admission the patient’s temperature was 38° C, hemoglobin was 86 g/L, and platelets were 34 × 109 per liter, with a leukoerythroblastic reaction and 27% large malignant lymphoid cells. LDH was 2266 U/L (normal, <210). The bone marrow histology and immunostaining were consistent with the diagnosis of intravascular lymphoma. The patient received six cycles of chemotherapy with the ACOP12 protocol. After a 15-month period of complete remission, the patient experienced a relapse and died 2 months later.
Case 3
A 66-year-old Chinese male, a recent immigrant from China, was referred for assessment for a 4-month history fever of unknown origin associated with anorexia and weight loss. Physical examination did not reveal any lymphadenopathy or hepatosplenomegaly. Peripheral blood count showed white blood cell count of 7.6 × 109 per liter, Hb of 90 g/L, and platelets 142 × 109/l. A bone marrow examination revealed hypercellularity with prominent hemophagocytosis. Further investigations unveiled hepatosplenomegaly by CT scan and LDH of 3092 IU. A liver biopsy was done, and the results were nondiagnostic. A punch biopsy of the skin did not show any lymphoma. Over a period of 3 weeks, the patient deteriorated with rising LDH (up to 16,295 U/L), severe anemia, and thrombocytopenia (platelet nadir = 7 × 109 per liter). Therapy with corticosteroid and intravenous gamma globulin was initiated. The patient responded initially but died 1 week later of cardiorespiratory failure. A repeat bone marrow was done the day before his demise (5 wks after initial biopsy) that showed marked hemophagocytosis and clusters of immature cells with prominent nucleoli. These cells were identified to be B lymphocytes with an intravascular location.
MATERIALS AND METHODS
Morphology and Immunophenotypic Analysis
Bone marrow aspiration samples and biopsy sections were processed routinely. Peripheral blood and bone marrow aspirate smears were stained with Giemsa. The bone marrow core biopsies were fixed in B5 after decalcification and stained routinely with hematoxylin and eosin (H&E). Immunohistochemistry on paraffin sections was performed on all cases using antibodies to CD20, CD3, BCL-2, and Factor VIII (DAKO, Glostrup, Denmark) using an automated immunostainer (Ventana Medical Systems, Tuscon, AZ). Positive and negative controls were run with the study cases and stained appropriately. Stains were considered positive when the majority of the tumor cells stained positively. Flow cytometric immunophenotyping was performed on Case 2 using routine three-color analysis on a Coulter Epics Profile II and a direct antibody labeling technique. Mouse monoclonal antibodies used included CD2, 3, 4, 5, 7, 8, 10, 11c, 14, 19, 20, 23, and 45 (Becton Dickinson, Mississauga, ON), goat anti-κ and anti-λ (Tago, Fort Wayne, IN) and FMC-7 (Silenus Laboratories, Boronia, VIC) labeled with phycoerythrin, fluorescein isothiocyanate or Cy5.
Standard and Molecular Cytogenetic Analysis
Chromosome analysis was performed on bone marrow aspirates using standard 24-hour culture and harvest procedures and G-banding as previously reported (39). The karyotypes are described according to ISCN 1995 (40).
M-FISH analysis was performed according to manufacturer protocols (Metasystems CmbH, Altlussheim, GER). Briefly, denaturation of slides and probes cocktails was conducted for 5 minutes at 75° C; hybridization was performed over 3 days. After hybridization, washing and posthybridization steps were performed. Metaphase preparations were then counterstained with DAPI and analyzed. To assess whether the duplication of 18q (Case 2) was associated with BCL2 gene amplification and whether the 14q32 breakpoint (Case 3) involves the IGH gene, FISH analysis using the LSI IGH/BCL2 probe was performed according to manufacturer protocol (VYSIS, Downers Grove, IL). Briefly, slides and probes cocktails were denatured for 5 minutes at 72° C, and hybridization was performed overnight. After hybridization, washing and posthybridization steps were performed. Metaphase preparations were then counterstained with DAPI and analyzed. M-FISH and FISH analysis were performed using a Zeiss microscope (Axioplan 2) equipped with the appropriate filters (DAPI, FITC, Spectrum Orange, TRITC, Cy5, DEAC) and the Metasystems ISIS imaging software programs.
RESULTS
Histopathology
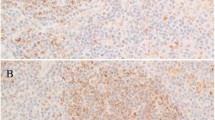
All three cases showed similar findings, with involvement of bone marrow sinuses by diffuse large B-cell lymphoma (DLBCL). The bone marrows were hypercellular, with small interstitial collections of large neoplastic lymphoma cells composed predominantly of centroblasts. These were scattered throughout the interstitial spaces in groups of 8 to 20 cells. Typically, the large B cells were surrounded by sinus lining cells, highlighted clearly by staining with anti-factor VIII. The malignant lymphoma cells stained strongly with anti-CD20 and also labeled with anti-BCL-2 in Case 2. The other two cases stained strongly with anti-CD20 but failed to express BCL-2 protein. A representative H&E section of Case 3 is shown in Figure 1A, and the accompanying CD20 stain, in Figure 1B. The bone marrow aspirate in Case 3 was characterized by marked hemophagocytosis involving predominantly erythrocytes. This was not a feature of the other two cases. Flow cytometric analysis of the bone marrow in Case 2 revealed a small population of abnormal cells expressing CD19, CD20, monoclonal surface λ, CD5, and CD11c but negative for CD23 and FMC-7.

A, representative histology of bone marrow H&E from Case 3, arrow showing dilated sinusoid with large atypical lymphoid cells. B, CD20 stain of the marrow from Case 2, showing clusters of positive-staining cells. CandD, G-band karyotypes from Case 2 and Case 3 (see Table 2 for karyotype description), m indicates the large marker chromosome. E, M-FISH karyotype from Case 2, arrows indicating der(10)t(10;22) and der(17)t(17;22), bracket indicating the duplication within chromosome 18. F, M-FISH karyotype from Case 3, arrows indicating the t(11;14) and der(14)t(11;14). GandH, FISH result with the specific probe for IGH (14q32) and BCL2 (18q23), showing duplication of the BCL2 signal on the duplicated 18 from Case 2 (g) and two copies of IGH signal, one on a normal 14 and one on the der(11)t(11;14) from Case 3 (h).
Cytogenetic Analysis
An abnormal karyotype was detected in each case by G-banding analysis (Table 2). Residual normal metaphases were detected in Case 1 only. The karyotypic changes in all cases were predominantly balanced and unbalanced structural rearrangements involving additions, deletions, duplication, inversions and translocations. A single-marker chromosome was identified in Case 3 (Figure 1d). M-FISH was used to further resolve the ambiguous karyotypic alterations. For Case 1, the abnormal clone was not detected by the M-FISH procedure. In Case 2 an add(1)(q42.1) was shown to be an intra-chromosomal inv(1) (q42q4?), the add(10)(p11.2) was shown to be a der(10)t(10;22)(p12;q?), the del(17)(p11.2p13) was shown to be a der(17)t(17;22)(p11;q?), and the dup(18)(q11q23) was shown to be a complex duplication of 18q (Figure 1e). For Case 3, the large marker chromosome was shown to consist entirely of chromosome 3 material, presumably resulting from a complex intra-chromosomal inversion. An ins(11;?)(q13;?) was shown to be a der(11)dup(11)(q?) resulting from internal 11q duplication. This der (11) was also involved in a cryptic balanced t(11;14)(p15;q32) (verified by FISH analysis, data not shown) to which the add(14)(q32) and +add(14)(q32) were related (Figure 1f). The M-FISH refined karyotype nomenclature is included in Table 2.
FISH analysis using a BCL2 probe revealed one normal copy on normal 18 and multiple copies on the derivative 18 (containing the 18q duplication) in Case 2 (Figure 1g). In Case 3 two normal BCL2 copies were seen (Figure 1h). No evidence of splitting of the IGH probe was identified in either case. In Case 3 with a balanced t(11;14), an intact IGH signal was indicative of a breakpoint proximal to the IGH locus at 14q32.3.
We undertook a band-by-band assessment of the chromosomal breakpoint sites and regions of deletion and duplication involved in the 14 previously reported IVL karyotypes (Table 3) and the 3 reported herein. Structural rearrangements with breakpoints near gene loci implicated in lymphoma were identified in a portion of the 17 IVL karyotypes. A breakpoint at 14q32 that might have involved the IGH locus was identified in four of the 11 AIVL cases. These alterations included an abnormal 14 reported as ?add(14)(q22–32), a der(14)t(1;14)(q2?;q32), an abnormal 14 reported as der(14)(q32;q15), and the t(11;14)(p15;q32) in Case 3 of this report. In the latter case, involvement of IGH gene was excluded. Breakpoints involving other lymphoma-associated oncogene loci including BCL6 (3q27), CMYC (8q24), PAX5 (9p13), BCL1 (11q13), and BCL2 (18q21) and BCL3 (19q13) were seen in 2, 1, 1, 3, 1, and 2 cases each, respectively.
Regions of duplication that were apparent in >20% of the karyotypes included all of chromosome 18 (+18) and the 11q13 → qter region. Trisomy 18 or duplication of 18q was present in 7 of 17 karyotypes. Recurrent deletions involved a number of chromosomes and chromosomal regions, including: −1, del(3)(q21–qter), −4, del(6)(q11–qter), del(8)(p21 → qter), del(9)(p21 → pter), del(9)(q22 → qter), −15, del(18)(q23) and del(22)(q22–qter). The most common recurrent deletion, seen in >50% of cases, was del(6)(q21–q23).
We assessed for similarities and differences in the identified breakpoints, deletions, and duplications seen in classical IVL versus the Asian variant, consisting in this group of 6 versus 11 reported karyotypes (Table 4). The two alterations that showed significant difference in frequency of involvement were deletion of 6q11–q13 and breakpoint at 6q11, seen more frequently in IVL than in AIVL (P = .03 and .03, respectively, Fisher’s exact test; Table 4).
DISCUSSION
Intravascular lymphoma (IVL) is a rare extranodal variant of DLBCL. Based on its morphological characteristics and clinical behavior it is considered to be a distinct subtype of DLBCL in the WHO classification (41). Accurate and timely diagnosis is still a problematic issue (42, 43). The majority of cases are diagnosed at autopsy. The clinical presentation is predominated by fever, neurological alterations, and skin lesions with absence of lymphadenopathy. Bone marrow involvement is rarely reported, but when present, the malignant cells are scant and confined to the bone marrow sinusoids (11). Hemophagocytosis has been described in few cases of IVL that mainly, but not exclusively (44, 45), involve Japanese patients (26). Murase et al. (27) suggested that this is a distinct variant of IVL and named it an Asian variant of intravascular large B-cell lymphoma (AIVL).
We have identified three cases of IVL. The clinicopathological characteristics of these cases are summarized in Table 1. The first patient had a “typical” presentation of IVL with predominantly dermatological and neurologic manifestations. In the second case, the presentation was unusual with prominent bone marrow involvement and circulating malignant cells. The clinical presentation of the third case was consistent with AIVL. We confirmed bone marrow involvement in all three cases by using anti-CD20 (L26) immunostain to highlight the malignant cells and factor VIII-related stain for delineating the vascular walls and confirming the intravascular pattern. IVL is a disseminated disease with simultaneous intravascular involvement of most body organs (including lung, heart, prostate, pancreas, kidney, testes, and adrenal glands) has been found repeatedly at autopsy (12). However, bone marrow involvement has been rarely reported in the literature (23). It has been suggested that the bone marrow involvement in IVL is underestimated as it is usually subtle and difficult to recognize by H&E staining only and the systematic use of immunohistochemistry could help identify additional cases (11, 23). DiGiuseppe et al. (46) detected monoclonal proliferation, by IgH gene rearrangement studies, in the bone marrow of 5/5 cases of IVL with apparently normal morphology and suggested that the regular application of this molecular technique would facilitate the detection of the marrow involvement in this disease.
From >250 cases of IVL published in the literature, karyotypic data have been reported in only 14 cases (4 classical and 10 Asian variant), and in all cases, complex cytogenetic abnormalities were found (Table 3). This low frequency of reported karyotypes is probably due to the diagnostic difficulties seen in this disease, because a large proportion of IVL cases is diagnosed at autopsy (42, 43). Another reason that may account for this low frequency is the unusual presentation of this tumor. Patients with IVL typically present with nonlocalizing neurological signs and symptoms and/or raised hyperpigmented or hemorrhagic skin lesions with absence of distinct tumoral mass or apparent involvement of hematopoietic organs (12). Usually, cytogenetic analysis is not performed as part of the initial work-up of these clinical manifestations. In three cases, clonal cytogenetic abnormalities were found in bone marrow (2 cases) and peripheral blood (1 case) samples, which showed apparently normal morphology (28, 29, 30). This suggests that peripheral blood and bone marrow involvement is probably underestimated by conventional morphology, and karyotyping of these sources of cellular material is worthwhile even in absence of apparent morphologic involvement.
To our knowledge, this is the first report to describe cytogenetic abnormalities in IVL analyzed by multicolor karyotyping. The application of M-FISH clarified the chromosomal abnormalities in the karyotypes of our reported cases. In all cases, standard G-banding showed chromosomal alterations with unidentified additional material. By M-FISH, cryptic translocations that include der(10)t(10;22), der(17)t(17;22), and balanced t(11;14) were identified. In Case 3, the additional material on 14q32 was revealed to be part of a balanced t(11;14)(p15; q32). The band 14q32 is known to contain the IGH gene. However, locus-specific FISH with an IGH probe did not show rearrangement of this gene, in this case suggesting a breakpoint proximal to 14q32.3. A review of the previously reported karyotypes revealed that breakpoints involving 14q32 locus were found in three other cases. Whether the translocation that we have found in this study is a unique or recurrent translocation in IVL will require cytogenetic analysis of additional cases supplemented by multicolor karyotyping, chromosome painting or telomeric FISH probes.
The quantitative analysis of our karyotypes and those previously published reveals that an overall pattern of chromosomal alterations typical of B-cell lymphomas and in particularly of DLBCL (38). However, recurrent translocations involving immunoglobulin loci and common oncogenes implicated in B-cell lymphoma (cyclin D1, BCL2, BCL6, MYC) have not been encountered. The most common recurrent alterations were −6/6q− (59%) and +18/dup(18q) (41%).
Complete or partial deletions of chromosome 6 were found in 10/17 (59%) karyotypes. The most commonly deleted region was located to 6q21–q23. Deletion of the long arm of chromosome 6 has been described in 14–31% of large series of NHL (47, 48) and has been associated with the full spectrum of this disease (49). By quantitative cytogenetic analysis, three commonly deleted regions have been found (48, 49). These are located at 6q21, 6q23, and 6q25. By loss of heterozygosity (LOH) studies, Gaidano et al. (50) found two distinct regions of minimal molecular deletion (RMD) at 6q25-q27 (RMD1) and at 6q21-q23 (RMD2). Offit et al. (48), comparing the regions of minimal molecular deletion with the common cytogenetic deletion regions, postulated the presence of a third region of minimal molecular deletion (RMD3) located at 6q23. Starostik et al. (51) confirmed this postulate by finding two LOH hot spots in gastric large cell lymphoma located at 6q21-q22.1 and 6q23.3-q25. In a recent study, Nanjangud et al. (38) observed, by using SKY techniques, that del(6q) was the most frequent deletion in DLBCL (38%) with 6q23, 6q21, 6q25–27, 6q15, and 6q11–13 being the most commonly deleted regions in a decreasing order of incidence. These results may suggest the presence of at least five tumor suppressor genes located on the long arm of chromosome 6 that could play roles in the pathogenesis of B-NHL. Moreover, correlation has been found between the distinct RMDs and the different subtypes of B-NHL (49). DLBCL has been associated with RMD2 located at 6q21–q23 (38, 49). We found that the commonly deleted region in IVL is located at 6q21–q23. These data indicate that IVL is morphologically, clinically, and probably genetically related to DLBCL.
Trisomy 18 occurs in 10–14% of NHL usually as a secondary abnormality (52, 53). Trisomy 18 or duplication of the long arm of chromosome 18 has been identified in 7/17 (41%) cases of IVL but not as a primary alteration. Gains of chromosome 18 material have been reported recently in DLBCL, follicular lymphoma, and MZL studied by comparative genomic hybridization (CGH) techniques (54, 55, 56). In these studies, it has been shown that the most commonly amplified region is mapped to bands 18q21–22. This region is known to contain the BCL2 oncogene at 18q21.3. BCL2 is implicated in the cellular apoptotic pathways. BCL2 overexpression leads to inhibition of apoptosis conferring a survival advantage to cells and contributes by this mechanism to the development of lymphoma. Monni et al. (55) demonstrated BCL2 oncogene amplification in all 6 cases of DLBCL, which showed gains in 18q by CGH. Dierlamm et al. (56) showed BCL2 protein overexpression in 6 of 7 cases of MZL, which had gains in 18q21. These data suggest an association between gains in 18q, BCL2 gene amplification, and BCL2 protein overexpression. From the three cases analyzed in this study, gain of 18q was detected in one case (Case 2). Locus-specific FISH with a BCL2 probe was applied in this case and showed one normal copy on normal 18 and prominent amplification on derivative 18 (Fig. 1E). When our three cases were examined by immunohistochemistry, only Case 2 showed BCL2 protein expression. BCL2 protein overexpression was systematically studied by immunohistochemistry in 13 cases of IVL and was positive in seven cases, but cytogenetic data were not available for these cases (5, 11, 57). Whether the over-expression of BCL2 in IVL is always associated with gain of 18q or could be due to other mechanisms cannot be answered presently. Identification of the normal cellular counterpart of IVL, studies of the BCL2 protein expression in these cases, and correlative studies between the overexpression of BCL2 and abnormalities in 18q are needed to enhance our understanding of the role of BCL2 in this unusual lymphoid neoplasm.
Although we have shown differences in the frequency of deletion of 6q11-q13 and breakpoint at 6q11 between IVL and AIVL, the number of cases available for assessment is still too small for reliable interpretation of these findings. The examination of additional karyotypic data using comprehensive molecular cytogenetic approaches (M-FISH and FISH) is needed to confirm our results and to assess whether more differences could be found between the two disease subtypes.
References
Ansell J, Bhawan J, Cohen S, Sullivan J, Sherman D . Histiocytic lymphoma and malignant angioendotheliomatosis: one disease or two. Cancer 1982; 50: 1506–1512.
Wrotnowski U, Milles SE, Cooper P . Malignant angioendotheliomatosis: an angiotropic lymphoma. Am J Clin Pathol 1985; 83: 244–248.
Sheibani K, Battifora H, Winberg CD, Burke JS, Ben-Ezra J, Ellinger GM, et al. Further evidence that “malignant angioendotheliomatosis” is an angiotropic cell lymphoma. N Engl J Med 1986; 314: 943–948.
Morikawa Y, Tohya K, Kuribayshi K, Saito K . A case of neoplastic angioendotheliomatosis is an angiotropic intravascular lymphoma. Appl Pathol 1989; 7: 322–328.
Bergmann M, Terzija-Wessel U, Blasius S, Kuchelmeister K, Kryne-Kubat B, Gerhard L, et al. Intravascular lymphomatosis of CNS: clinicopathological study and search for expression of oncoproteins and Epstein-Barr virus. Clin Neurol Neurosurg 1994; 96: 236–243.
Perniciaro C, Winkelemann RK, Daoud MS, Su WPD . Angiotropic intravascular lymphoma: immunohistochemical, ultrastructural and molecular genetic studies. Am J Dermatopathol 1995; 17: 242–248.
Elner VM, Hidayat AA, Charles NC, Davitz MA, Smith ME, Burgess D, et al. Neoplastic angioendotheliomatosis: a variant of malignant lymphoma: immunohistochemical and ultrastructural observations of three cases. Ophthalmology 1986; 93: 1237–1245.
Stroup RM, Sheibani K, Moncada A, Purdy LJ, Battifora H . Angiotropic (intravascular) large cell lymphoma: a clinicopathological study of seven cases with unique clinical presentation. Cancer 1990; 66: 1781–1788.
Torenbeek R, Scheltens Ph, Strack Van Schijndel RJ, Algra PR, Heimans JJ, Van der Valk P . Angiotropic intravascular large cell lymphoma with massive cerebral extension. J Neurol Neurosurg Psychiatry 1993; 56: 914–916.
Sepp N, Schuler G, Romani N, Geissler D, Gattringer C, Burg G, et al. “Intravascular lymphomatosis” (angioendotheliomatosis): evidence for a T-cell origin in two cases. Hum Pathol 1990; 21: 1051–1058.
Estallila OC, Koo CH, Brynes RK, Medeiros LJ . Intravascular large B-cell lymphoma: a report of five cases initially diagnosed by bone marrow biopsy. Am J Clin Pathol 1999; 112: 248–255.
Glass J, Hochberg FH, Miller DC . Intravascular lymphomatosis: a systemic disease with neurologic manifestations. Cancer 1993; 71: 3156–3164.
DiGiuseppe JA, Nelson WG, Seifter EJ, Boitnott JK, Mann RB . Intravascular lymphoma: a clinicopathological study of ten cases and assessment of response to chemotherapy. J Clin Oncol 1994; 12: 2573–2579.
Calamia KT, Miller A, Shuster EA, Perniciaro C, Menke DM . Intravascular lymphomatosis: a report of ten patients with central nervous system involvement and a review of the disease process. Adv Exp Med Biol 1999; 37: 249–265.
D’Agati V, Sablay LB, Knowles DM, Walter L . Angiotropic large cell lymphoma (intravascular malignant lymphomatosis) of the kidney. Hum Pathol 1989; 20: 263–268.
Yousem SA, Colby TV . Intravascular lymphomatosis presenting in the lung. Cancer 1990; 65: 349–353.
Dermier T, Dail DH, Aboulafia DM . Four varied cases of intravascular lymphomatosis and literature review. Cancer 1994; 73: 1738–1745.
Bogomolski-Yahlom V, Lossos IS, Okun E, Sherman Y, Lossos A, Polliack A . Intravascular lymphomatosis—an indolent or aggressive entity? Leuk Lymphoma 1998; 29: 585–593.
Suzumiya J, Ohshima K, Kanda M, Kato A, Shuda K, Kimura N, et al. Intravascular lymphomatosis associated with hypoalbuminemia. Leuk Lymphoma 1998; 32: 179–182.
Walls JG, Hong YG, Cox JE, McCabe KM, O’Brien KE, Allerton JP, et al. Pulmonary intravascular lymphomatosis; presentation with dyspnea and air trapping. Chest 1999; 115: 1207–1209.
Fulling KH, Gersell DJ . Neoplastic angioendotheliomatosis: histologic, immunohistochemical and ultrastructure findings in two cases. Cancer 1993; 51: 1107–1118.
Ip M, Chan KW, Chan IKL . Systemic inflammatory response syndrome in intravascular lymphomatosis. Intensive Care Med 1997; 23: 783–786.
Tucker TJ, Bardales RH, Miranda RN . Intravascular lymphomatosis with bone marrow involvement. Arch Pathol Lab Med 1999; 123: 952–956.
Emura I, Naito M, Wakabayshi M, Yoshizawa H, Arakawa M, Chou T . Detection of circulating tumor cells in a patient with intravascular lymphomatosis: a case study examined by cytology method. Pathol Int 1998; 48: 63–66.
Murase T, Nakamura S . An Asian variant of intravascular lymphomatosis: an updated review of malignant histiocytosis-like B-cell lymphoma. Leuk Lymphoma 1999; 33: 459–473.
Murase T, Nakamura S, Kawauchi K, Matsuzaki H, Sakai C, Inaba T, et al. An Asian variant of intravascular large B-cell lymphoma: clinical, pathological and cytogenetic approaches to diffuse large B-cell lymphoma associated with haemophagocytic syndrome. Br J Haematol 2000; 111: 826–834.
Murase T, Nakamura S, Tashiro K, Suchi T, Hirga J, Hayasaki N, et al. Malignant histiocytosis-like B-cell lymphoma, a distinct pathologic variant of intravascular lymphomatosis: a report of five cases and review of the literature. Br J Haematol 1997; 99: 656–664.
Molina A, Lombard C, Donlon T, Bangs CD, Dorfman F . Immunohistochemical and cytological studies indicate that malignant angioendotheliomatosis is a primary intravascular (angiotropic) lymphoma. Cancer 1990; 66: 474–479.
Davey DD, Munn R, Smith LW, Cibull ML . Angiotrophic lymphoma: presentation in uterine vessels with cytogenetic studies. Arch Pathol Lab Med 1990; 114: 879–882.
Nakahara T, Saito T, Muroi A, Sugiura Y, Ogata M, Sugiyama Y, et al. Intravascular lymphomatosis presenting as an ascending cauda equina: conus medullaris syndrome: remission after weekly CHOP therapy. J Neurol Neurosurg Psychiatry 1999; 67: 304–306.
Shimazaki C, Inaba T, Shimura K, Okamoto A, Takahashi R, Hirai H, et al. B-cell lymphoma-associated with hemophagocytic syndrome: a clinical, immunological and cytogenetic study. Br J Haematol 1999; 104: 672–679.
Ichikawa N, Kobayashi H, Ito T, Hiroshi S . Chromosomal aberrations after stimulation with lipopolysaccharide in a patient with Asian variant intravascular lymphomatosis. Cancer Genet Cytogenet 2000; 120: 83–84.
Tsukadaira A, Okubo Y, Ogasawara H, Urushibata K, Honda T, Miura I, et al. Chromosomal aberrations in intravascular lymphomatosis. Am J Clin Oncol 2002; 25: 178–181.
Veldman T, Vignon C, Schrock E, Rowley JD, Ried T . Hidden chromosome abnormalities in haematological malignancies detected by multicolor spectral karyotyping. Nat Genet 1997; 15: 406–410.
Sawyer JR, Lukas JL, Munshi N, Raman Desikan K, Singhal S, Mehta J, et al. Identification of new nonrandom translocations in multiple myeloma with multicolor spectral karyotyping. Blood 1998; 92: 4269–4278.
Rowley JD, Reshmi S, Carlson S, Roulston D . Spectral karyotype analysis of T-cell acute leukemia. Blood 1999; 93: 2038–2042.
Mohr B, Bornhauser M, Thiede C, Schakel U, Illmer T, Pascheberg U, et al. Comparison of spectral karyotyping and conventional cytogenetics in 39 patients with acute myeloid leukemia and myelodysplastic syndrome. Leukemia 2000; 14: 1031–1038.
Nanjangud G, Rao PH, Hegde A, Teruya-Feldstein J, Donnelly G, Qin J, et al. Spectral karyotyping identifies new rearrangements, translocations, and clinical associations in diffuse large B-cell lymphoma. Blood 2002; 99: 2554–2561.
Horsman DE, Gascoyne RD, Coupland RW, Coldman A, Adomat SA . Comparison of cytogenetic analysis, Southern analysis and polymerase chain reaction for the detection of t(14;18) in follicular lymphoma. Am J Clin Pathol 1995; 103: 472–478.
Mitelman F, editor. Basel, Switzerland: Karger; 1995. ISCN. An international system for human cytogenetic nomenclature.
Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink K, Vardiman J, et al. World Health Organization of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the clinical advisory committee meeting. J Clin Oncol 1999; 17: 3835–3849.
Delvin T, Moll S, Hulette C, Morgenlander JC . Intravascular malignant lymphomatosis with neurologic presentation: factors facilitating antemortem diagnosis. South Med J 1998; 91: 672–676.
Vieren M, Sciot R, Robberecht W . Intravascular lymphomatosis of the brain: a diagnostic problem. Clin Neurol Neurosurg 1999; 101: 33–36.
Dufau JP, Le Tourneau A, Molina T, Le Houcq M, Claessens YE, Rio B, et al. Intravascular large B-cell lymphoma with bone marrow involvement at presentation and haemophagocytic syndrome: two Western cases in favour of a specific variant. Histopathology 2000; 37: 509–512.
Viali S, Hutchinson DO, Hawkins TE, Croxson MC, Thomas M, Allen JP, et al. Presentation of intravascular lymphomatosis as lumbosacral polyradiculopathy. Muscle Nerve 2000; 23: 1295–1300.
DiGiuseppe JA, Hartmann DP, Freter C, Cossman J, Mann RB . Molecular detection of bone marrow involvement in intravascular lymphomatosis. Mod Pathol 1997; 10: 33–37.
Offit K, Chaganti RS . Chromosomal aberrations in non-Hodgkin’s lymphoma. Hematol Oncol Clin North Am 1991; 5: 853–869.
Offit K, Parsa NZ, Gaidano G, Fillipa DA, Louie D, Pan D, et al. 6q deletions define clinico-pathological subsets of non-Hodgkin’s lymphoma. Blood 1993; 82: 2157–2162.
Johansson B, Mertens F, Mitelman F . Cytogenetic evolution patterns in non-Hodgkin’s lymphoma. Blood 1995; 86: 3905–3914.
Gaidano G, Hauptschein RS, Parsa NZ, Offit K, Rao PH, Lenoir G, et al. Deletions involving two distinct regions of 6q in B-cell non-Hodgkin lymphoma. Blood 1992; 80: 1781–1787.
Starostik P, Greiner A, Schultz A, Zettl A, Peters K, Rosenwald A, et al. Genetic aberrations in gastric high-grade B-cell lymphoma. Blood 2000; 95: 1180–1187.
Armitage JO, Sanger WG, Weisenburger DD, Harrington DS, Linder J, Bierman PJ, et al. Correlation of secondary cytogenetic abnormalities with histologic appearance in non-Hodgkin’s lymphoma bearing t(14;18)(q32;q21). J Natl Cancer Inst 1988; 80: 576–580.
Konishi K, Sakurai M, Nakao H, Maseki N, Kaneko Y, Yagiri Y, et al. Chromosome abnormalities in malignant lymphoma in patients from Kurashiki: histological and immunophenotypic correlations. Cancer Res 1990; 50: 2698–2703.
Bentz M, Werner CA, Dohner H, Barth TFE, Siebert R, Schroder M, et al. High incidence of chromosomal imbalances and gene amplifications in the classical variant of follicle center lymphoma. Blood 1996; 88: 1437–1444.
Monni O, Joensuu H, Franssila K, Knuutila S . DNA copy number changes in diffuse large B-cell lymphoma-comparative genomic hybridization study. Blood 1996; 87: 5269–5278.
Dierlamm I, Rosenberg C, Stul M, Pittaluga S, Wlodarska I, Michaux L, et al. Characteristic pattern of chromosomal gains and losses in marginal zone B cell lymphoma detected by comparative genomic hybridization. Leukemia 1997; 11: 747–758.
Ponzoni M, Arrigoni G, Gould VE, Del Curto B, Maggioni M, Scapinello A, et al. Lack of CD29 (β1 Integrin) and CD54 (ICAM-1) adhesion molecules in intravascular lymphomatosis. Hum Pathol 2000; 31: 220–226.
Acknowledgements
This study was supported in part by the National Institutes of Health, Research Grant 1-UO1-CA84967–01.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Khoury, H., Lestou, V., Gascoyne, R. et al. Multicolor Karyotyping and Clinicopathological Analysis of Three Intravascular Lymphoma Cases. Mod Pathol 16, 716–724 (2003). https://doi.org/10.1097/01.MP.0000077515.68734.85
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000077515.68734.85
Keywords
This article is cited by
-
Intravascular lymphoma: look through the small vessels
Annals of Hematology (2017)
-
Tandem triplication of the BCL2 gene in CD5-positive intravascular large B cell lymphoma with bone marrow involvement
Annals of Hematology (2014)
-
NRG1 gene rearrangements in clinical breast cancer: identification of an adjacent novel amplicon associated with poor prognosis
Oncogene (2005)
