
Abstract
Purpose
Reanalysis of exome sequencing data when results are negative may yield additional diagnoses. We sought to estimate the contribution of clinical geneticists to the interpretation of sequencing data of their patients.
Methods
The cohort included 84 probands attending a tertiary genetics institute (2015–2018) with a nondiagnostic result on clinical exome sequencing performed in one of five external laboratories. The raw data were uploaded to the Emedgene bioinformatics and interpretation platform for reanalysis by a team of two clinical geneticists, the geneticist directly involved in the patient’s care, and a bioinformatician.
Results
In ten probands (11.9%), a new definitive diagnosis was reached based on genes that were known to be associated with the phenotype at the time the original report was issued. The main reasons for a negative exome result were incorrect interpretation of the clinical context and absence of OMIM entry. Pathogenic variants in genes with previously unknown gene–disease associations were discovered to be causative in three probands. In total, new diagnoses were established in 13/84 individuals (15.5%).
Conclusion
Direct access to complete clinical data and shortening of time to including gene–phenotype associations in databases can assist the analytics team and reduce the need for additional unnecessary tests.
Similar content being viewed by others


INTRODUCTION
The interpretation of exome sequencing data can be complex, and the identification of relevant disease-causing variants remains an ongoing challenge. Negative results may be due to technological limitations (noncoding or expansion mutations, incomplete coverage, uniparental disomy, large indels, chromosomal rearrangements, and copy-number variants); unknown gene–disease associations; and epigenetic, multifactorial, or nongenetic factors (toxins, oxygen deprivation, premature delivery).1 Previous studies have reported that data reanalysis may offer an additional diagnostic yield of 10–15%, mostly owing to newly published gene–disease associations or identification of small deletions or duplications following copy-number variant analysis.2,3 An additional cause of analytics failure is incomplete recognition of the patient’s phenotype.4,5
The aim of the present study was to estimate the contribution of clinical geneticists to the interpretation of the exome sequencing data of their patients.
MATERIALS AND METHODS
Study setting and participants
The study was conducted at the Raphael Recanati Genetic Institute of Rabin Medical Center, a major tertiary hospital in central Israel. According to departmental policy, samples are sent to an external laboratory for clinical exome sequencing when the clinical geneticist suspects an as-yet undiagnosed monogenic disorder in an individual without a clinical diagnosis or a highly heterogeneous condition that may be caused by mutations in several different genes. The institute uses five external laboratories: four are located in the United States or Europe and are accredited by the College of American Pathologists (CAP) and registered or certified through CLIA, and one is located in Israel (outside our hospital) and is accredited by the Israel Ministry of Health.
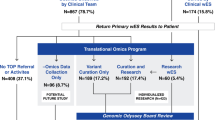
From 2015 to 2018, clinical exomes from 114 probands followed at the Recanati Genetic Institute were sent for analysis to an external diagnostic laboratory, and the raw sequencing data were received in return. Exome sequencing was not covered by the Israel National Health Insurance, and the patients paid privately for the tests and for the return of the raw data. In 30/114 cases, the molecular diagnosis was established. In the remaining 84 cases, the result was nondiagnostic, and written consent was obtained from the patients to conduct a reanalysis of the raw sequencing data by the local team. Data reevaluation by the local team was carried out on the same set of samples tested by the external laboratories. Samples from additional family members were used for confirmation and segregation analysis with Sanger sequencing. Phenotypic information was derived by direct review of the electronic health records, which included the pedigree; the results of clinical, laboratory, and radiographic investigations; and the facial images of the participants. All study participants received updated genetic counseling.
The study protocol was approved by the Rabin Medical Center research ethics committee.
Exome sequencing interpretation
The FASTQ files of the individuals with a nondiagnostic clinical exome result were uploaded to the Emedgene bioinformatics and interpretation platform (Emedgene Technologies, Ltd, Mazor, Israel; http://emedgene.com/) for reanalysis by a team of two clinical geneticists and one bioinformatician at our institute. Following mapping (using the BWA-MEM algorithm) and variant calling (using multiple algorithms), low quality and polymorphic variants were filtered out. The Emedgene platform provides suggestions of most likely candidate variants and various options for variant filtering by inheritance mode, quality, frequency, and severity. The list of candidate genes focused on missense, nonsense, frameshift, and splicing variants with a minor allele frequency of <1% in multiple populations (based on 1000 Genomes, ESP 6500, ExAC, gnomAD, and a local database). The variant effect was determined by Polyphen, SIFT, MutationTaster, LRT, GERP, SiPhy, PhastCons, and single-nucleotide variant (SNV) score. Candidate variants with the relevant phenotypic association were further discussed by the local team. Incidental/secondary findings were not assessed.
During the reevaluation process, the team reviewed the phenotypic data of each patient with the geneticist directly involved in the patient’s care, and the facial images were examined. A variant was considered causative if it was classified as pathogenic or likely pathogenic6 and if the geneticist managing the care of the family judged the disorder to be clearly compatible with the phenotype. In one case, a variant of unknown significance was suggested as a strong candidate in trans with a likely pathogenic variant. Following data reanalysis of proband-only exomes, candidate variant segregation in the parents and additional family members was performed by Sanger sequencing.
RESULTS
The characteristics of the 84 probands with a nondiagnostic clinical exome sequencing result are summarized in Table 1. Most of the cohort were children, and the most frequent indication for exome sequencing was neurodevelopmental disorder. A new definitive diagnosis related to genes that had been known to cause human disorders at the time the original laboratory report was issued was reached in 10/84 patients (11.9%) (cases 1–10, Table 2). None of the causative variants had appeared in the publicly available databases during the time from the first to the repeated comprehensive analysis. In all but one case, the suggested variants in genes with known gene–disease associations were classified as pathogenic or likely pathogenic. In one proband, a variant of unknown significance (in trans with a likely pathogenic variant) was suggested as a strong candidate (case 1, Table 2). In this child, blood analysis revealed extremely high total bile salt levels (up to 588 μmol/L, normal level 0.0–10), a finding compatible with the suggested diagnosis. In addition, in three probands (cases 11–13), variants in genes with previously unknown gene–disease associations were discovered to be causative (Table 2); these cases were subsequently published.7,8,9 In 12 probands, the causative variant identified had not been mentioned in any of the sections of the original laboratory report. In case 11, a candidate variant in POC5 was listed in the original laboratory report as a variant of unknown significance. In total, diagnoses were established in 13/84 individuals (15.5%) for genes with both previously known and unknown involvement in human disease. In seven cases, the mode of inheritance was autosomal dominant: in six, the variant appeared de novo, and in one, the variant was inherited from the mother. Four cases represented autosomal recessive inheritance, and two disorders were related to genes on the X chromosome. The candidate variants were confirmed by Sanger sequencing in cases 1, 2, 3, 4, 5, 6, 8, 9, 11,12, and 13. In cases 7 and 10, depth of coverage, mapping, and base and genotype quality were high.
DISCUSSION
When a nondiagnostic result is obtained on exome sequencing, additional tests may be applied, such as genome sequencing, genome-wide deletion evaluation, and RNA sequencing.10,11,12 Some authors report that genome sequencing offers limited diagnostic utility compared with reanalysis of exome sequencing.13 In the present study, many solved cases involved mutations in genes with a known genotype–disease relationship at the time of the original analysis. All the causative variants suggested by the local team were seen by the evaluating team in the external laboratories; in only one case (case 3) was the causative variant considered to be an artifact. In 4/10 cases (cases 7, 8, 9, 10), the reason for the negative exome result was incorrect interpretation of the clinical context. When the diagnostic laboratories were contacted with the result of the reevaluation, they explained that they had noted the variant but did not have sufficient confidence it was related to the phenotype. In cases 7 and 10, long-term developmental observation might have contributed to successful identification of the causative variant. Despite laboratory efforts to make test requisitions user-friendly, many specimens are accompanied by insufficient clinical information and the clinical data provided are frequently insufficient to successfully prioritize variants in genes causing similar but nevertheless different phenotypes. The team evaluating the data in external laboratories often has no access to such important clinical features as age at onset of the disorder, progressive versus nonprogressive disease course, typical gestalt, growth parameters, age at onset and types of seizures, response to treatment of seizures, and, very importantly, patient images.
Additional explanations for negative result were absence of OMIM entry for gene–phenotype association and lack of inclusion of the most relevant publications in the existing OMIM entry (3/10 cases). In cases 1 and 5, no entry was present in OMIM, but convincing gene–phenotype associations were reported in the literature by the time of the negative exome report. In case 6, an intragenic deletion in the NFIA gene was reported as causing structural brain abnormalities and abnormal development on 22 January 2014 (PMID: 24462883), and a truncating pathogenic variant was reported on 26 February 2015 (PMID: 27081522); the laboratory report was issued on 29 July 2015. The OMIM entry on the disorder related to the NFIA gene was created on 8 February 2011 but updated only on 31 October 2017. In one case (case 4), incorrect judgment regarding mode of inheritance was made and in one case (case 2) we could not understand the cause for missing the causative variant because the disorder was listed in OMIM at the time of the original report.
Because most genomic testing is outsourced, clinical geneticists provide little input on variant prioritization. In addition, they are not always part of the team interpreting the data in external laboratories. Data interpretation may be further complicated by the possibility of novel clinical features of a known disorder or the presence of more than one monogenic disorder in a single individual.14 Obviously, variant definition should not serve as a substitute for expert clinical opinion on the possible role of the variant in causing the patient’s phenotype.
We conclude that the increased diagnostic yield in this study was mostly related to two factors: (1) access of the local analytic team to more detailed phenotypic data and images of the patients and the possibility to estimate the changing phenotype over time, and (2) lack of inclusion of gene–disease association or relevant publications in OMIM at the time of the original report. This study shows that local reanalysis of exome sequencing data can increase the diagnostic yield, thereby reducing the need for additional costly and unnecessary tests such as genome sequencing. When clinical exome sequencing analysis is performed in a hospital-based diagnostic laboratory where clinical geneticists are members of the diagnostic team, automating the analysis and using an intuitive, user-friendly variant filtration interface can greatly assist clinicians in interpretation of the findings. When exome sequencing analysis is performed by an external laboratory, better communication between the laboratory and the clinical team, for example via an interactive web-based platform, can improve diagnostic accuracy. Such platforms can serve as a virtual meeting point between the clinician and the laboratory for rapid and efficient data management, by (1) clarifying specific gene coverage, (2) adding phenotypic details, (3) exchanging written communication between persons involved in data evaluation, (4) reporting variant segregation results, and (5) reporting a change in variant classification. It is important to note that medicolegal aspects should be taken into account and discussed before implementing this type of platform in routine medical care. In addition, shortening the time to inclusion of gene–phenotype associations and updating entries in databases can significantly improve variant interpretation.
References
Lelieveld SH, Spielmann M, Mundlos S, Veltman JA, Gilissen C. Comparison of exome and genome sequencing technologies for the complete capture of protein-coding regions. Hum Mutat. 2015;36:815–822.
Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19:209–214.
Xiao B, Qiu W, Ji X. Marked yield of re-evaluating phenotype and exome/target sequencing data in 33 individuals with intellectual disabilities. Am J Med Genet A. 2018;176:107–115.
Aarabi M, Sniezek O, Jiang H. Importance of complete phenotyping in prenatal whole exome sequencing. Hum Genet. 2018;137:175–181.
Pena LDM, Jiang YH, Schoch K. Looking beyond the exome: a phenotype-first approach to molecular diagnostic resolution in rare and undiagnosed diseases. Genet Med. 2018;20:464–469.
Richards S, Aziz N, Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Weisz Hubshman M, Broekman S, van Wijk E. Whole-exome sequencing reveals POC5 as a novel gene associated with autosomal recessive retinitis pigmentosa. Hum Mol Genet. 2018;27:614–624.
Orenstein N, Goldberg-Stern H, Straussberg R. A de novo GABRA2 missense mutation in severe early-onset epileptic encephalopathy with a choreiform movement disorder. Eur J Paediatr Neurol. 2018;22:516–524.
Zeharia A, Friedman JR, Tobar A, Saada A, Konen O, Fellig Y. Mitochondrial hepato-encephalopathy due to deficiency of QIL1/MIC13 (C19orf70), a MICOS complex subunit. Eur J Hum Genet. 2016;24:1778–1782.
Belkadi A, Bolze A, Itan Y. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A. 2015;112:5473–5478.
Gambin T, Akdemir ZC, Yuan B. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017;45:1633–1648.
Cummings BB, Marshall JL, Tukiainen T. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 2017;9:eaal5209. https://doi.org/10.1126/scitranslmed.aal5209. Accessed on 25 October 2018.
Alfares A, Aloraini T, Subaie LA. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet Med. 2018 March 22; https://doi.org/10.1038/gim.2018.41 [Epub ahead of print]. Accessed on 25 October 2018.
Posey JE, Harel T, Liu P. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31.
Acknowledgements
The manuscript was edited by the Medical Editing Office of Rabin Medical Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Basel-Salmon, L., Orenstein, N., Markus-Bustani, K. et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet Med 21, 1443–1451 (2019). https://doi.org/10.1038/s41436-018-0343-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0343-7
Keywords
This article is cited by
-
Phenotypic compatibility and specificity in genomic variant classification
European Journal of Human Genetics (2024)
-
Systematic reanalysis of genomic data by diagnostic laboratories: a scoping review of ethical, economic, legal and (psycho)social implications
European Journal of Human Genetics (2024)
-
Ordering genetic testing by neurologists: points to consider
Journal of Neurology (2023)
-
Reanalysis of whole-exome sequencing (WES) data of children with neurodevelopmental disorders in a standard patient care context
European Journal of Pediatrics (2023)
-
Skewed X-chromosome inactivation in unsolved neurodevelopmental disease cases can guide re-evaluation For X-linked genes
European Journal of Human Genetics (2023)
