
Abstract
Epigenetics, and especially DNA methylation, have recently become provocative biological explanations for early-life environmental effects on later health. Despite the large increase in papers on the topic over the last few years, many questions remain with regards to the biological feasibility of this mechanism and the strength of the evidence to date. In this review, we examine the literature on early-life effects on epigenetic patterns, with special emphasis on social environmental influences. First, we review the basic biology of epigenetic modification of DNA and debate the role of early-life stressful, protective, and positive environments on gene-specific, system-specific, and whole-genome epigenetic patterns later in life. Second, we compare the epigenetic literatures of both humans and other animals and review the research linking epigenetic patterns to health in order to complete the mechanistic pathway. Third, we discuss physical environmental and social environmental effects, which have to date, generally not been jointly considered. Finally, we close with a discussion of the current state of the area’s research, its future direction, and its potential use in pediatric health.
Similar content being viewed by others

Main
There is a large body of research that examines the link between the social environment (i.e., individual and community-level characteristics (1)) and health. More recent has been the rapid increase in work focusing on epigenetics, and especially DNA methylation, as a mechanism by which the social environment can affect health. Strong evidence suggests that the social environment modifies important epigenetic patterns that play a role in later health, wellbeing, and behavior (2,3,4). This review describes some of the recent work in this area, but first reviews DNA methylation (its biological functions and measurement) and then outlines some of the research hurdles and opportunities.
Biology of Methylation
The epigenome refers to all heritable information that does not originate in the sequence of DNA. In this context, we use “heritable” to refer to information that is transmissible to daughter cells during cellular replication rather than to transgenerational inheritance. Within the epigenome are histone modifications (acetylation, methylation, and others), DNA methylation, and possibly noncoding RNA. The human body is composed of at least 400 different cellular types, and despite having the same DNA, different cell types have distinct gene expression (mRNA) patterns (5). The major effector of cellular diversity and differentiation is the regulation of mRNA transcription; epigenetic modification of DNA and histones is a key modulator of this process. One potentially exciting property of the epigenome is that, unlike the DNA sequence, it may be modified by environmental and pharmacological interventions, providing the potential for reversing the effect of adverse life events on later health and wellbeing (6).
Recall that DNA is organized as a linear polymeric molecule, in which the four nucleotides (deoxyadenosine triphosphate, A; thymidine triphosphate, T; deoxyguanosine triphosphate, G; deoxycytosine triphosphate, C) form the DNA polymer through a phosphodiester bond. The flow of information in a cell (according to the Central Dogma) goes from DNA to messenger RNA (mRNA), to protein. Genes are arrayed along chromosomes, and a gene can be viewed as consisting of the arrangement of base sequences that specifies a complementary mRNA, and, therefore, a specific protein, together with those nearby DNA sequences that determine when and to what extent the gene is transcribed into RNA.
At the 5′ end of the gene is a sequence of bases termed the promoter. Binding of these proximal control elements by a series of transcription factors activates the process by which RNA polymerase synthesizes a complementary strand of mRNA. After the primary RNA copy of the gene is synthesized, significant processing takes place. Following export from the nucleus of the cell, the mRNA is engaged by a ribosome and used as a template for synthesis of a polypeptide chain.
The regulation of mRNA transcription is a key mechanism by which the cell regulates the amount and nature of mRNA and proteins to meet the requirements of specific cell types and cell functions.
Changes in cellular functions occur though biochemical modification of protein structure (phosphorylation, enzymatic cleavage, other modifications). In addition, upon receipt of an appropriate signal, the cell can deploy or withdraw specific transcription factors within minutes, thereby rapidly modulating the mRNA transcription of specific genes and hence the expression of their protein products. This type of signaling response is rapid, and easily reversible. Epigenetic changes to DNA also modulate transcription by altering chromatin structure and transcription factor accessibility. Changes in DNA methylation are generally construed to be stable and to exert effects over a sustained period. Recently, this view has been supplemented by studies that provide evidence for rapid alterations in DNA methylation in response to signaling events in the central nervous system. Of particular relevance to this discussion, a growing literature identifies changes in DNA methylation as critical to synaptic plasticity, learning, and neurogenesis (7). Further, neuronal activation has been associated with rapid changes in DNA methylation, and with associated changes in mRNA expression (8).
DNA Methylation
Many regions of genes, including their promoters, enhancers, and silencers contain dinucleotides consisting of cytosine alternating with guanine (abbreviated CpG). Areas in which the proportion of CpG’s is greater than statistically predicted are termed CpG islands. Wherever a CpG occurs, the C is susceptible to being modified by one of a number of DNA methyltranferases through the addition of a methyl (CH3) group (forming 5-methylcytosine, 5mC) (9). 5-methylcytosine is successively oxidized by the ten-eleven translocation (Tet) family proteins to form 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (10,11,12) ( Figure 1 ). Thymine DNA glycosylase can excise 5fC or 5caC and replace it with unmethylated cytosine by base excision repair (13). Standard bisulfite-based approaches for detecting methylated cytosine do not distinguish between 5mC and 5hmC, and until recently this had limited the field’s appreciation of the role of 5hmC in moderating gene expression.

Deoxycytidine modification. Cytosine is methylated at the 5’ carbon by DNMT to form 5mC. It is successively oxidized by TET proteins to form 5hmC, 5fcC, and 5caC. For simplicity, only the products of the first two modifications are shown. DNMT, DNA methyltransferases; TET, Ten-eleven translocation.
The promoters of many housekeeping genes contain CpG islands, and methylation of these sites attenuates or halts transcription. Since the addition of a methyl group to cytosine is a covalent reaction, it may be an enduring change; furthermore, the DNA replication apparatus has mechanisms for ensuring that the corresponding CpG is methylated in daughter cells, helping to assure a stable phenotype across generations of cells.
Clusters of variably methylated CpG residues are found not only in promoters, but also interspersed within genes, along intergenic regions and within silencers and enhancers. The role played in cellular physiology by methylation of these other CpG sites is the subject of considerable research, and they may be more important in controlling transcription of many genes than the CpG islands. In addition to other mechanisms for regulating RNA expression, noncoding RNA transcripts also alter gene expression by DNA methylation regulation; for example, by forming a triplex with DNA and recruiting DNA methyltransferases to silence rRNA genes (14) or activation of specific genes by inhibiting DNA methyltransferase through direct interaction (15).
The methylation of DNA is just one way in which a cell can create an epigenetic mark. DNA is tightly coiled around highly basic proteins called histones. One effect of this winding is to greatly compress the DNA, allowing it to be packaged into a cell nucleus. Fully extended, the DNA of a chromosome would extend about 75 mm but in its compact (heterochromatin) state it is about 5 µm. Often, when DNA is tightly wound on a nucleosome, DNA regulatory sites (such as the promoter) become inaccessible to transcription signals, and the affected genes become silent. Histone proteins have several sites at which they can be covalently modified, principally by methylation or acetylation. The effect of histone modification may be to unwind the DNA, thereby freeing regulatory sites for interactions with various transcriptional activator proteins. In addition, histone marks may directly affect transcription, through interactions with regulatory proteins. As is the case with DNA methylation, the cell is able to duplicate the histone marks on newly synthesized histones that are destined for daughter cells. Thus, histone-based epigenetic marks are heritable even though they are not coded in the DNA.
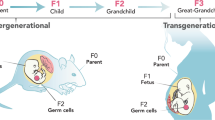
While epigenetic marks are heritable from parent cell to daughter cell, this does not entail that in multicellular organisms, such as humans, most epigenetic marks are transferred directly from parent to child. During the process of gamete formation epigenetic marks are cleared; imprinted genes are remethylated prior to fertilization, and each generation has been thought to develop a new set of epigenetic marks in response to environmental signals and associated with differentiation. There are two well-established mechanisms by which epigenetic marks can be “transferred” to a new generation. First, epigenetic signals that modify parental behavior or physiology can affect the postnatal environment of the offspring, shaping their behavior through epigenetic effects. Second, an environmental signal delivered to a pregnant female can simultaneously affect the epigenetic patterns of three generations: the pregnant female (F0), the fetus (F1), and the germ line cells of the F1 fetus (F2). Neither of these is an example of true transgenerational epigenetic inheritance, although both of these mechanisms may be very important for the health of succeeding generations.
The first mechanism is illustrated by the work of Weaver, Meaney, et al. (16), who showed that variations in maternal care can serve as the basis for an epigenetic behavioral transmission of individual differences in stress reactivity across generations, probably through epigenetic programming of the expression of hippocampal growth hormone receptors in the offspring (reviewed in refs. (17,18)). Notably, the epigenetic, gene expression, and hypothalamic-pituitary-adrenal stress axis effects could be pharmacologically reversed by infusion of a drug that affects histone acetylation, lending further support to the concept that this epigenetically-induced change in expression of the glucocorticoid receptor in response to maternal behavior is related to changes in chromatin structure and is causal. While suggestive, because the agent used to reverse methylation in this study (a histone deacetylase (HDAC) inhibitor) induces generalized demethylation (16), it is not possible to formally ascribe reversion of phenotype to changes at a particular CpG locus; proof that epigenetic changes at a specific CpG residue moderate specific behavioral or physiological traits still awaits demonstration. The second mechanism is reflected in the effects of diethylstilbestrol (DES) administration to pregnant woman. The woman who received DES to prevent miscarriage did not benefit in this regard (F0), but their in utero daughters (F1) developed clear cell adenocarcinoma of the vagina and a number of other serious effects (19). The daughters’ in utero (F1) sons had an increased incidence of hypospadias (20); the daughters’ sons (the grandsons, F2) may have an increased risk of hypospadias (21) and there is some evidence of infertility in F2 woman, although this remains to be confirmed (22). While it is possible that the multiple generation effects observed with DES are a valid example of transgenerational epigenetic inheritance through the germ lines, establishing this would require either that a fourth generation be affected (F3) or that the offspring of DES sons (the F2 generation of males) demonstrate DES-related effects.
There is now some limited evidence for transgenerational epigenetic inheritance of environmentally induced traits through germ line transmission of DNA methylation and histone marks. For example, Carone, et al reported that paternal diet affected promoter methylation and mRNA expression of hepatic genes in the offspring (23); this report provides a potential biological mechanism for the otherwise puzzling observation that early paternal smoking is associated with an increase in the sons’ weight (24). Supporting the idea that epigenetic patterning can be transmitted by sperm to succeeding generations, Brunner et al. (25) observed marking of sperm by post-translational modification of histones and their associated protamines. Others have provided a theoretical and experimental context in which histone and DNA signals can be transgenerationally transmitted (26,27,28). Even so, the role of true transgenerational inheritance remains quite unclear in general, and to our knowledge, studies to examine the potential role of transgenerational inheritance related to social stress have not been reported.
As discussed, at the biochemical level, epigenetics affects transcription and ultimately the protein repertoire of a cell. Epigenetic mechanisms serve four essential cellular roles: (i) X-chromosome inactivation; (ii) differentiation; (iii) imprinting; (iv) medium and long-term transcriptional control (29). Typically methylation of the promoter region is associated with decreased gene expression, while methylation in other areas (e.g., intergenetic region) is associated with both increased and decreased gene expression (9,30). This review focuses on how social and environmental signals shape DNA methylation, presumably through medium and long-term transcriptional control. Imprinting, which is underlies parent of origin inheritance, is based on the differential DNA methylation of genes present on paternal or maternal chromosomes. For example, transmission of a particular deletion on chromosome 15 (q 11–13) from the mother produces Angelman syndrome, while transmission of a similar deletion from the father produces Prader-Willi syndrome, a much different phenotype (31). Genes such as IGF2 and IGF2R are imprinted (32), and are also known to be epigenetically modified under certain conditions of prenatal adversity. This leaves open the possibility, as yet untested, that imprinting (i.e., parent of origin effects) plays a role transmission of social or external environmental stress from parent to child. Since sperm are imprinted at Igf2 (32), this also implies that male transmission of stress-induced epigenetic effects are possible (see below). Aberrations in DNA methylation are frequently associated with cancer, but this phenomenon is not within the scope of this review.
Evidence for Social Environmental Effects on Methylation
Animal Literature
To date, the largest literature on environmental effects on methylation is from animal studies and there are many reviews (2,4,17,27,33,34,35,36,37,38). In general, the findings suggest that early life stressors significantly modify several epigenetic marks in multiple genes and regions. For example, evidence from animal studies has shown that maternal exposure to drugs, stress, and toxicants can alter epigenetic gene programming in the brain and contribute to neurodevelopmental and behavioral deficits in the offspring (34,39,40). Similarly, maternal separation and maternal stress have been studied to investigate mechanisms of long-lasting effect of stress on adult health and wellbeing as well as their mediation by epigenetic processes (2,4,37,38,41). Animals subjected to early postnatal stress display a range of behavioral alterations depending on the type of stressors and their duration (4,17). For example, in brain tissue, DNA methylation was found to be altered following postnatal stress in genes regulating the hypothalamic-pituitary-adrenal stress axis: Nr3c1, Avp, Crh, and Crh2 (16,42,43,44) and neurotrophic genes: BDNF, 5HT1A, and Gad1 (45,46,47). Many of these changes were even observable in adulthood (41). As a specific example, mice that were exposed to chronic and unpredictable maternal separation from postnatal day 1 to 14 were differentially methylated in several candidate genes, including: MeCP2, CB1, and CRFR2 (44).
The importance of 5hmC, in gene by environment interactions is only beginning to be appreciated. 5hmC-containing DNA has been shown to be enriched in the brain (48), and is important for neuronal differentiation and pluripotent stem cell reprogramming (reviewed in ref. (49)). In accordance with a role in gene regulation, genome-wide localization experiments show that 5-hmC is associated with enhancers and gene bodies (50,51). Recent studies showed that fear extinction in mice results in genome-wide accumulation of 5-hmC in the infralimbic prefrontal cortex (52). Together with the work on memory, plasticity, and environmental signaling that was cited previously, this body of research provides tantalizing clues to how environmental signals may shape both the development of the brain through both rapid and long-term modifications of the central nervous system epigenome.
More recent is the work showing the effect of positive environments on epigenetic patterns. Exposure to stimulating social environments in early life induces epigenetic modifications that influence expression of brain-derived neurotropic factor (BDNF) in adult mice, probably by inducing acetylation of histone residues related to the BDNF gene (53). Integrating positive early life measures is one avenue we believe could have great promise.
Human Literature
Among the early life social predictors for methylation profiles, child abuse and neglect have received the largest attention in relation to epigenetic patterns (17,54). For example, childhood maltreatment was first shown to influence methylation patterns of the glucocorticoid receptor gene (NR3C1) in the brain (55), but has also been shown to be evident in other tissues as well (17,56,57,58,59). Indeed recent work suggests that childhood maltreatment may result in many epigenetic changes—into the thousands of CpG sites (60). However, even broader parenting behaviors, and not just the most extreme, have shown evidence of influencing methylation (4). In fact, the NR3C1 gene is one of the most studied in the literature, with over 40 papers to date—most showing the strong effect of abuse and neglect and a relatively reduced but still discernable effect of other psycho-social measures (18).
There are several other phenotypes, environments, and epigenetic loci that have been examined (2,4,17,27,33,34,35,36,37,38,40,61). Here, we provide just a few to demonstrate the range of findings. For example, maternal psychopathology, criminal behaviors, and substance use was associated with greater OXTR methylation at birth (62) This might result in reduced prosocial and bonding behavior later in life (63). A broad range of maternal stressors in infancy and paternal stressors in childhood influences methylation of several genes (MKRN1, TRDN, PGAM2, PKN1, PGAM2) in adolescence (64). Mother’s folic acid intake before and during pregnancy resulted in differential insulin-like growth factor 2 (IGF2) methylation and expression (65). Increased serotonin transporter gene (SERT, SLC6A4) DNA methylation is associated with bullying victimization (66).
Prenatal maternal stress affects placental function, and it is likely that in part, these effects are moderated by changes in placental methylation patterns. The long-term effects of epigenetically moderated placenta dysfunction on health and behavior of children is clearly moderated by postnatal parenting choices, and this interaction is a rich subject for additional study (39). Even broader measures of the social environment such as neighborhoods and larger social groups appear to have some influence on methylation patterns, probably by effects on stress, although to what extent these are permanent changes is still under investigation (18,34,35,41,57,67,68,69,70).
Developmental Timing of Environment Effects
Most of the research on social environmental effects on epigenetics does not explicitly compare the sequence and duration of epigenetic effects, which limits the ability to propose developmentally relevant models. Typically studies do not analyze more than one time period, but a broader look at the whole research area provides some hint at potential developmental differences. For example, in utero environments have received a substantial focus (2,34). Fetal programing—or the developmental origins of adult health and disease—has thus become a major focus of epigenetic work (71). There is substantial evidence that in utero environments appear to have the largest effects on later phenotypes (33,65,72,73,74,75,76). Sensitivity to environmental influence is observed in later time periods (77,78)—not as significantly as during the fetal period. However, it is important to note that even in adulthood methylation can change; for example social isolation in adult mice modified epigenetic patterns for HDAC1, HDAC3, and SLC6A4 (79).
Social vs. Physical Environment
Although we have focused mostly on social environments, we have mentioned a few studies from the much larger physical environment (physical, chemical, and biological agent exposure (1)) and methylation literature (4,72,80,81). For example, the physical environment is the primary focus of the majority of studies of the in utero environment, examining measures such as heavy metal exposure, pollution, maternal and neonatal diet, and other toxicant exposure (36,74,75,76). Interestingly, the social and physical environments are not often studied together despite there being substantial evidence they are correlated and may even interact (36,81).
Epigenetics and Children’s Health
Animal Literature
The literature linking DNA methylation to early life health is significantly smaller, but growing rapidly when compared to the literature on early life events (34,80). Most often this work models methylation as a mediator between the social environment and health. For example, variations in maternal care are tied to behavioral differences in rats; the glucocorticoid receptor gene (NR3C1) showed changes in methylation associated with the maternal care and later behavior (16,55). Similarly, neonatal handling influences hippocampal glucocorticoid receptors in adult rats through epigenetic modification of NR3C1 (82). Other behavioral phenotypes are also being studied, such as learning and learned behavior (83). There are a few more direct studies of methylation on early life phenotypes. For instance, methylation of 5HT1A, which codes the 5-HT1A receptor seems to affect influences serotonergic signaling, altering social anxiety and social recognition memory (44).
Human Literature
Like the animal literature, most work using methylation has a limited focus for pediatric outcomes—mostly through mental health and stress reactivity. Most work on mental health and epigenetics is in adults, but some work involving children is beginning to emerge (84,85). A growing literature shows that epigenetic patterns are related to depression and depressive-like symptoms, but these are often cross-sectional studies (86,87). In one study, OXTR methylation at birth was associated with callous and unemotional traits, but not internalizing behaviors, later in adolescence (62). Epigenetic changes in ID3, GRIN1, and TPPP genes may confer risk for depression in maltreated children (88). According to the authors of this study, each of these genes is plausibly tied to stress and neural development, and changes in the expression of these genes could contribute to subsequent depression.
Developmental programing of the hypothalamic-pituitary-adrenal stress axis is another major focus of the epigenetics to pediatric health research (89). This includes effects on stress reactivity as well as broader psychopathology (67,77). For instance, increased serotonin transporter gene (SERT, SLC6A4) DNA methylation is associated with blunted cortisol response to stress in childhood (66). Further, methylation patterns appear to influence stress resilience throughout childhood development (61,90).
Measuring DNA Methylation
Determining whether a specific CpG site is methylated is a relatively straightforward application of sequencing technology or the polymerase chain reaction (PCR), and several approaches are in routine laboratory use. More recently, chip technology has been applied to determine the methylation state of 500,000 to 1 million CpG sites per DNA sample (91,92,93). The advent of hybridization methylation assays has permitted the field to move beyond candidate locus studies to epigenome-wide surveys (84). In addition, the larger arrays have allowed for other uses of the methylation data including estimating cell type distributions and methylation-determined biological age (94,95). A caveat to these technologies is that the common practice of bisulfite conversion, which deaminates unmethylated cytosine to uracil, does not distinguish between 5-methylcytosine and 5-hmC. Fortunately, a number of technologies have been developed to distinguish these modifications (reviewed in ref. (96)) and some are compatible for use in larger arrays (97).
Hurdles and Future Direction
Although the array measurement technology is straightforward, early experience suggests that there are significant challenges to analyzing the data, ranging from batch effects (artifacts induced by minor day to day variation in lab procedures) to the statistical and visualization challenges implicit in very large numbers of repeated measures in a limited number of samples. Furthermore, while it is relatively straightforward to identify which CpG sites are hyper- or hypo-methylated under a certain condition, it is much more difficult to associate this observation with a specific functional significance. The biological effect of a change in methylation status at a particular CpG or cluster of CpGs is often unknown or unpredictable based on our current understanding of the “methylation code.” While work with both rodents and humans has demonstrated the value of methylation changes in explaining how environmental inputs are translated to durable behavioral effects, the best examples of this sort of work have, so far, depended upon measurement of the methylation state of specific sites with known or clearly predictable functions (45,98,99). How methylation state profiles across hundreds of thousands of sites should be correlated with underlying social inputs and health or behavior states remains an important topic for research. There is not yet a sufficiently large body of research using array technology to understand how reproducible these measurements and conclusions will be across sample sets, or whether signals detected with hybridization arrays will routinely be validated with sequencing-based approaches. Since the methylation state of differentiated tissues is highly specific, it is not clear how methylation profiles developed in circulating blood cells or saliva cells will provide information about changes in DNA from less accessible tissue such as brain, cells of the autonomic nervous system, or specific immune cells. This later point is a major challenge for the field that could be confronted thorough relatively straightforward experiments in mice and postmortem observations in humans.
Since the putative mechanism by which epigenetic changes such as DNA methylation affect phenotype is through modulating RNA expression, it is important to correlate methylation changes and mRNA levels. In addition, DNA methylation-induced changes in miRNA expression have been observed in several model systems, including cancer, but have not been examined in the context of social or behavioral epigenetics (100). Are perturbations in DNA methylation associated with corresponding mRNA expression changes in the relevant genes? For cancer-related methylation abnormalities, there is a productive literature that indicates that this is sometimes (but not always) the case (101,102). Studies have also demonstrated that maternal protein restriction (in rats) is associated with DNA methylation changes and relevant modulation of mRNA expression in the offspring. Of interest, folic acid supplementation attenuated both changes (103).
An important function of DNA methylation and other epigenetic marks is to confer cell-specific gene expression identity that is formed during embryonic development (4,92,104). There is some evidence to suggest that saliva is both easier and potentially more closely related to brain methylation patterns than blood (92). However, changes in those tissues’ methylation patterns have not been thoroughly examined. Furthermore, even if the specific cell types’ methylation profiles were perfectly or predictably correlated, it is important to recall that methylation signatures are usually drawn from a mixture of cell types. This means that fluctuations in cell type distribution can be a confounding variable (77,105). Studies that can account for cell distribution should do so whether with estimated cell distributions (95) or cells counted (e.g., via flow cytometry), with appropriate adjustments.
Will measuring epigenetic changes provide a target for intervening to attenuate the health effects of environmental stressors (70,106)? We have made reference to the observation that folic acid supplementation reverses changes in DNA methylation (and RNA expression) associated with maternal protein restriction (103). If this approach can be translated to humans, would it be possible to intervene in a targeted way to ameliorate the fetal effects of maternal environmental or social stress? There is presently very little data to aid in evaluating the possibility of this sort of approach. Therapy designed to correct abnormalities in DNA methylation may be effective and is under clinical trial investigation for particular forms of cancer (such as mantle cell leukemia) that are characterized by significant abnormalities in DNA methylation (107). It is possible that these approaches will be extended to other perturbations of epigenetics, including those imposed by adverse physical or social environments, but our understanding of how this could work is still rudimentary. It is much more likely that in the near future alterations in DNA methylation will serve as a biomarker or intermediate outcome measures for established treatments (105). For example, Okada (108) found in depressed individuals that the level of promoter methylation of the 5-HTT gene (SLC6A) predicted magnitude of response to therapy, and BDNF methylation status could be a proxy marker for previous suicidal attempts and a clinical biomarker for poor treatment outcomes of suicidal ideation in depression (109).
While a great deal of excitement surrounds the insight that social and external environments produce robust adverse effects in offspring through epigenetic mechanisms, the field still requires considerable maturation (3,110). A longitudinal study design with multiple time points of methylation data is important to assess the kinetics and direction of epigenetic changes, but this sort of data is unusual in human studies (62). Making this issue more trenchant is that sensitivity to environmental effects is likely to vary during development and maturation (2). Similarly, sample sizes are typically very small and not representative of any particular population. While epigenetic research has taught us a great deal about the mechanisms that underlie the interplay between environment, genetic endowment, and child health and wellbeing, the field is still very young, and there are likely to be many surprises.
Summary
Epigenetic modifications, such as histone modifications and DNA methylation, are important in cellular differentiation and development because they enable cells with identical DNA to elicit differential transcriptional programs and adapt to environmental cues.
Environmental conditions such as maternal stress and parenting behavior affect DNA methylation during fetal development and throughout life.
Stress-associated behavioral phenotypes and psychopathology are correlated with altered DNA methylation of several genes, including regulators of the hypothalmic-pituitary-adrenal axis.
Although the connection between the environmental impact on the epigenome and child health and well being is only beginning to be deciphered, DNA methylation may someday be useful as a therapeutic target or biomarker to quantify or predict treatment response.
Statement of Financial Support
Funding was provided by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01 HD076592).
References
Health Disparities: Linking Biological and Behavioral Mechanisms With Social and Physical Environments. National Institutes of Health: Bethesda, MD, 2000.
Champagne FA. Epigenetic influence of social experiences across the lifespan. Dev Psychobiol 2010;52:299–311.
Meloni M, Testa G. Scrutinizing the epigenetics revolution. Biosocieties 2014;9:431–56.
Szyf M. DNA methylation, behavior and early life adversity. J Genet Genomics 2013;40:331–8.
Razin A. CpG methylation, chromatin structure and gene silencing-a three-way connection. EMBO J 1998;17:4905–8.
Miller GE, Chen E, Fok AK, et al. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc Natl Acad Sci USA 2009;106:14716–21.
Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci 2010;13:1319–23.
Guo JU, Ma DK, Mo H, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 2011;14:1345–51.
Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009;462:315–22.
Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010;466:1129–33.
Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011;333:1300–3.
Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–5.
He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011;333:1303–7.
Schmitz KM, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 2010;24:2264–9.
Di Ruscio A, Ebralidze AK, Benoukraf T, et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013;503:371–6.
Weaver IC, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004;7:847–54.
Anacker C, O’Donnell KJ, Meaney MJ. Early life adversity and the epigenetic programming of hypothalamic-pituitary-adrenal function. Dialogues Clin Neurosci 2014;16:321–33.
Turecki G, Meaney MJ. Effects of the social environment and stress on glucocorticoid receptor gene methylation: a systematic review. Biol Psychiatry 2014. (doi:10.1016/j.biopsych.2014.11.022)
Hoover RN, Hyer M, Pfeiffer RM, et al. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N Engl J Med 2011;365:1304–14.
Klip H, Verloop J, van Gool JD, Koster ME, Burger CW, van Leeuwen FE ; OMEGA Project Group. Hypospadias in sons of women exposed to diethylstilbestrol in utero: a cohort study. Lancet 2002;359:1102–7.
Brouwers MM, Feitz WF, Roelofs LA, Kiemeney LA, de Gier RP, Roeleveld N. Hypospadias: a transgenerational effect of diethylstilbestrol? Hum Reprod 2006;21:666–9.
Titus-Ernstoff L, Troisi R, Hatch EE, et al. Menstrual and reproductive characteristics of women whose mothers were exposed in utero to diethylstilbestrol (DES). Int J Epidemiol 2006;35:862–8.
Carone BR, Fauquier L, Habib N, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010;143:1084–96.
Pembrey ME, Bygren LO, Kaati G, et al.; ALSPAC Study Team. Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet 2006;14:159–66.
Brunner AM, Nanni P, Mansuy IM. Epigenetic marking of sperm by post-translational modification of histones and protamines. Epigenetics Chromatin 2014;7:2.
Campos EI, Stafford JM, Reinberg D. Epigenetic inheritance: histone bookmarks across generations. Trends Cell Biol 2014;24:664–74.
Gudsnuk K, Champagne FA. Epigenetic influence of stress and the social environment. ILAR J 2012;53:279–88.
Braun K, Champagne FA. Paternal influences on offspring development: behavioural and epigenetic pathways. J Neuroendocrinol 2014;26:697–706.
Hedges DJ, Deininger PL. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat Res 2007;616:46–59.
Bird AP. CpG-rich islands and the function of DNA methylation. Nature 1986;321:209–13.
Zucchi FC, Yao Y, Metz GA. The secret language of destiny: stress imprinting and transgenerational origins of disease. Front Genet 2012;3:96.
Bartolomei MS. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev 2009;23:2124–33.
Billack B, Serio R, Silva I, Kinsley CH. Epigenetic changes brought about by perinatal stressors: a brief review of the literature. J Pharmacol Toxicol Methods 2012;66:221–31.
Kundakovic M. Prenatal programming of psychopathology: the role of epigenetic mechanisms. J Med Biochem 2013;32:313–24.
Lewis CR, Olive MF. Early-life stress interactions with the epigenome: potential mechanisms driving vulnerability toward psychiatric illness. Behav Pharmacol 2014;25:341–51.
Mirbahai L, Chipman JK. Epigenetic memory of environmental organisms: a reflection of lifetime stressor exposures. Mutat Res Genet Toxicol Environ Mutagen 2014;764-765:10–7.
Peña CJ, Bagot RC, Labonté B, Nestler EJ. Epigenetic signaling in psychiatric disorders. J Mol Biol 2014;426:3389–412.
Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol 2011;31:363–73.
Monk C, Spicer J, Champagne FA. Linking prenatal maternal adversity to developmental outcomes in infants: the role of epigenetic pathways. Dev Psychopathol 2012;24:1361–76.
Jensen CL, Champagne FA. Epigenetic and neurodevelopmental perspectives on variation in parenting behavior. Parent Sci Pract 2012;12:202–11.
Provençal N, Binder EB. The effects of early life stress on the epigenome: From the womb to adulthood and even before. Exp Neurol 2015;268:10–20.
Murgatroyd C, Spengler D. Polycomb binding precedes early-life stress responsive DNA methylation at the Avp enhancer. PLoS One 2014;9:e90277.
Elliott E, Ezra-Nevo G, Regev L, Neufeld-Cohen A, Chen A. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat Neurosci 2010;13:1351–3.
Franklin TB, Russig H, Weiss IC, et al. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry 2010;68:408–15.
Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci 2010;30:13130–7.
Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry 2009;65:760–9.
Franklin TB, Linder N, Russig H, Thöny B, Mansuy IM. Influence of early stress on social abilities and serotonergic functions across generations in mice. PLoS One 2011;6:e21842.
Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009;324:929–30.
Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 2014;156:45–68.
Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol 2011;12:R54.
Szulwach KE, Li X, Li Y, et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet 2011;7:e1002154.
Li X, Wei W, Zhao QY, et al. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci USA 2014;111:7120–5.
Branchi I, Karpova NN, D’Andrea I, Castrén E, Alleva E. Epigenetic modifications induced by early enrichment are associated with changes in timing of induction of BDNF expression. Neurosci Lett 2011;495:168–72.
De Bellis MD, Zisk A. The biological effects of childhood trauma. Child Adolesc Psychiatr Clin N Am 2014;23:185–222, vii.
McGowan PO, Sasaki A, D’Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 2009;12:342–8.
Perroud N, Paoloni-Giacobino A, Prada P, et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Transl Psychiatry 2011;1:e59.
Lutz PE, Turecki G. DNA methylation and childhood maltreatment: from animal models to human studies. Neuroscience 2014;264:142–56.
Romens SE, McDonald J, Svaren J, Pollak SD. Associations between early life stress and gene methylation in children. Child Dev 2015;86:303–9.
Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 2008;3:97–106.
Yang BZ, Zhang H, Ge W, et al. Child abuse and epigenetic mechanisms of disease risk. Am J Prev Med 2013;44:101–7.
Waters CS, Hay DF, Simmonds JR, van Goozen SH. Antenatal depression and children’s developmental outcomes: potential mechanisms and treatment options. Eur Child Adolesc Psychiatry 2014;23:957–71.
Cecil CA, Lysenko LJ, Jaffee SR, et al. Environmental risk, Oxytocin Receptor Gene (OXTR) methylation and youth callous-unemotional traits: a 13-year longitudinal study. Mol Psychiatry 2014;19:1071–7.
Swain JE, Konrath S, Brown SL, et al. Parenting and Beyond: Common Neurocircuits Underlying Parental and Altruistic Caregiving. Parent Sci Pract 2012;12:115–23.
Essex MJ, Boyce WT, Hertzman C, et al. Epigenetic vestiges of early developmental adversity: childhood stress exposure and DNA methylation in adolescence. Child Dev 2013;84:58–75.
Hoyo C, Murtha AP, Schildkraut JM, et al. Methylation variation at IGF2 differentially methylated regions and maternal folic acid use before and during pregnancy. Epigenetics 2011;6:928–36.
Ouellet-Morin I, Wong CC, Danese A, et al. Increased serotonin transporter gene (SERT) DNA methylation is associated with bullying victimization and blunted cortisol response to stress in childhood: a longitudinal study of discordant monozygotic twins. Psychol Med 2013;43:1813–23.
Gudsnuk KM, Champagne FA. Epigenetic effects of early developmental experiences. Clin Perinatol 2011;38:703–17.
Toyokawa S, Uddin M, Koenen KC, Galea S. How does the social environment ‘get into the mind’? Epigenetics at the intersection of social and psychiatric epidemiology. Soc Sci Med 2012;74:67–74.
Tung J, Gilad Y. Social environmental effects on gene regulation. Cell Mol Life Sci 2013;70:4323–39.
Turecki G, Ota VK, Belangero SI, Jackowski A, Kaufman J. Early life adversity, genomic plasticity, and psychopathology. Lancet Psychiatry 2014;1:461–6.
Gluckman PD, Hanson MA, Buklijas T, Low FM, Beedle AS. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol 2009;5:401–8.
Kaminen-Ahola N, Ahola A, Maga M, et al. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet 2010;6:e1000811.
Guo L, Choufani S, Ferreira J, et al. Altered gene expression and methylation of the human chromosome 11 imprinted region in small for gestational age (SGA) placentae. Dev Biol 2008;320:79–91.
Perera F, Tang WY, Herbstman J, et al. Relation of DNA methylation of 5’-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS One 2009;4:e4488.
Pilsner JR, Hall MN, Liu X, et al. Influence of prenatal arsenic exposure and newborn sex on global methylation of cord blood DNA. PLoS One 2012;7:e37147.
Pilsner R, Hu H, Ettinger A, et al. Influence of prenatal lead exposure on genomic methylation of cord blood DNA. Environ Health Perspect 2009;117:1466–71.
Guintivano J, Kaminsky ZA. Role of epigenetic factors in the development of mental illness throughout life. Neurosci Res 2014 (doi:10.1016/j.neures.2014.08.003).
Heim C, Binder EB. Current research trends in early life stress and depression: review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp Neurol 2012;233:102–11.
Siuda D, Wu Z, Chen Y, et al. Social isolation-induced epigenetic changes in midbrain of adult mice. J Physiol Pharmacol 2014;65:247–55.
Champagne FA. Interplay Between Social Experiences and the Genome: Epigenetic Consequences for Behavior. In: Sokolowski MB, Goodwin SF, eds. Gene-Environment Interplay. Elsevier Academic Press Inc: San Diego, 2012. pp. 33–57.
Kundakovic M, Gudsnuk K, Franks B, et al. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc Natl Acad Sci USA 2013;110:9956–61.
Champagne FA. Early environments, glucocorticoid receptors, and behavioral epigenetics. Behav Neurosci 2013;127:628–36.
Dias BG, Maddox SA, Klengel T, Ressler KJ. Epigenetic mechanisms underlying learning and the inheritance of learned behaviors. Trends Neurosci 2015;38:96–107.
Klengel T, Pape J, Binder EB, Mehta D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology 2014;80:115–32.
Provencal N, Binder EB. The neurobiological effects of stress as contributors to psychiatric disorders: focus on epigenetics. Curr Opin Neurobiol 2015;30:31–7.
Dalton VS, Kolshus E, McLoughlin DM. Epigenetics and depression: return of the repressed. J Affect Disord 2014;155:1–12.
Ignácio ZM, Réus GZ, Abelaira HM, Quevedo J. Epigenetic and epistatic interactions between serotonin transporter and brain-derived neurotrophic factor genetic polymorphism: insights in depression. Neuroscience 2014;275:455–68.
Weder N, Zhang H, Jensen K, et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J Am Acad Child Adolesc Psychiatry 2014;53:417–24.e5.
Griffiths BB, Hunter RG. Neuroepigenetics of stress. Neuroscience 2014;275:420–35.
Zannas AS, West AE. Epigenetics and the regulation of stress vulnerability and resilience. Neuroscience 2014;264:157–70.
Greenleaf WJ. Assaying the epigenome in limited numbers of cells. Methods 2015;72:51–6.
Non AL, Thayer ZM. Epigenetics for anthropologists: An introduction to methods. Am J Hum Biol 2015;27:295–303.
Michels KB, Binder AM, Dedeurwaerder S, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods 2013;10:949–55.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol 2013;14:R115.
Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012;13:86.
Plongthongkum N, Diep DH, Zhang K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat Rev Genet 2014;15:647–61.
Field SF, Beraldi D, Bachman M, Stewart SK, Beck S, Balasubramanian S. Accurate measurement of 5-methylcytosine and 5-hydroxymethylcytosine in human cerebellum DNA by oxidative bisulfite on an array (OxBS-array). PLoS One 2015;10:e0118202.
Francis D, Diorio J, Liu D, Meaney MJ. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science 1999;286:1155–8.
Liu D, Diorio J, Tannenbaum B, et al. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science 1997;277:1659–62.
Suzuki H, Maruyama R, Yamamoto E, Kai M. DNA methylation and microRNA dysregulation in cancer. Mol Oncol 2012;6:567–78.
Hinoue T, Weisenberger DJ, Lange CP, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012;22:271–82.
Selamat SA, Chung BS, Girard L, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res 2012;22:1197–211.
Altobelli G, Bogdarina IG, Stupka E, Clark AJ, Langley-Evans S. Genome-wide methylation and gene expression changes in newborn rats following maternal protein restriction and reversal by folic acid. PLoS One 2013;8:e82989.
Stankiewicz AM, Swiergiel AH, Lisowski P. Epigenetics of stress adaptations in the brain. Brain Res Bull 2013;98:76–92.
Mikeska T, Craig JM. DNA methylation biomarkers: cancer and beyond. Genes (Basel) 2014;5:821–64.
Fass DM, Schroeder FA, Perlis RH, Haggarty SJ. Epigenetic mechanisms in mood disorders: targeting neuroplasticity. Neuroscience 2014;264:112–30.
Ghai V, Sharma K, Abbi KK, Shimko S, Epner EM. Current approaches to epigenetic therapy for the treatment of mantle cell lymphoma. Adv Exp Med Biol 2013;779:257–66.
Okada S, Morinobu S, Fuchikami M, et al. The potential of SLC6A4 gene methylation analysis for the diagnosis and treatment of major depression. J Psychiatr Res 2014;53:47–53.
Kang HJ, Kim JM, Lee JY, et al. BDNF promoter methylation and suicidal behavior in depressive patients. J Affect Disord 2013;151:679–85.
Haig D. Commentary: The epidemiology of epigenetics. Int J Epidemiol 2012;41:13–6.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Mitchell, C., Schneper, L. & Notterman, D. DNA methylation, early life environment, and health outcomes. Pediatr Res 79, 212–219 (2016). https://doi.org/10.1038/pr.2015.193
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.193
This article is cited by
-
Epigenetic analyses in forensic medicine: future and challenges
International Journal of Legal Medicine (2024)
-
Prenatal vitamin intake in first month of pregnancy and DNA methylation in cord blood and placenta in two prospective cohorts
Epigenetics & Chromatin (2022)
-
SHOX2 methylation in Vietnamese patients with lung cancer
Molecular Biology Reports (2022)
-
The epigenetic etiology of cardiovascular disease in a longitudinal Swedish twin study
Clinical Epigenetics (2021)
-
Diagnosis of pulmonary nodules by DNA methylation analysis in bronchoalveolar lavage fluids
Clinical Epigenetics (2021)
