
Abstract
Mitochondrial antiviral signaling (MAVS) is a key adaptor in cellular antiviral innate immunity. We previously identified poly(C)-binding protein 2 (PCBP2) as a feedback inhibitor of MAVS that facilitates its degradation after viral infection, but little is known about the regulatory potential of poly(C)-binding protein 1 (PCBP1), which highly resembles PCBP2. Here we report that PCBP1 mediates housekeeping degradation of MAVS using the same mechanism as PCBP2 employs. Overexpression of PCBP1 impairs MAVS-mediated antiviral responses, while knockdown of PCBP1 exerts the opposite effect. The suppression is due to PCBP1-induced MAVS degradation. We observe that PCBP1 and PCBP2 show synergy in MAVS inhibition, but their expression patterns are distinct: PCBP1 is stably and abundantly expressed, while PCBP2 shows low basal expression with rapid induction after infection. Individual knockdown and subcellular fractionation analyses reveal that unlike the postinfection inhibitor PCBP2, PCBP1 continuously eliminates cellular MAVS. Our findings unravel a critical role of PCBP1 in regulating MAVS for both fine-tuning the antiviral immunity and preventing inflammation.
Similar content being viewed by others


Introduction
Cellular antiviral innate immunity involves the detection of viral double-stranded RNA by cytoplasmic receptors including retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) 1, 2, which interact with the adaptor mitochondrial antiviral signaling (MAVS, also known as virus-induced signaling adaptor/interferon (IFN)-β promoter stimulator 1/caspase activation and recruitment domain adaptor inducing IFN-β) 3, 4, 5, 6. MAVS contains an N-terminal caspase activation and recruitment domain required for receptor binding and a C-terminal transmembrane domain (TM) that targets the protein to the mitochondria and peroxisomes 7, 8. At the receptor-proximal level, MAVS serves as a nexus to integrate the signaling of RIG-I-like receptors (RLRs) and mediate the downstream TANK-binding kinase 1 (TBK1)/IκB kinase ɛ activation 9. These kinases liberate transcription factors such as nuclear factor κB (NF-κB) and IFN regulatory factor 3/7, leading to the induction of type I IFNs and pro-inflammatory cytokines 10, 11. Once secreted, type I IFNs rapidly induce multiple IFN-stimulated genes 12, 13 that orchestrate an antiviral state in the cells 14. Due to its prominent role in propagating antiviral signaling 15, 16, MAVS is subject to meticulous modulations by different proteins 17, which fine-tune the cellular responses over the course of viral infection 18, 19, 20, 21.
Previously, we identified both PCBP1/PCBP2; also known as α-complex protein 1/2 or heterogeneous nuclear ribonucleoprotein E1/2 as MAVS-interacting candidates in a yeast two-hybrid screen, and delineated a novel cascade for MAVS degradation by the PCBP2-AIP4 (Atrophin 1-interacting protein 4, also known as Itchy homolog E3 ubiquitin ligase) axis 22. Of note, PCBP1 is highly homologous to PCBP2 and frequently collaborates with PCBP2 in various cellular processes 23, 24, 25, 26, 27. However, it is still unknown whether PCBP1 holds an impact on cellular antiviral immunity.
Here we report that PCBP1 negatively modulates cellular antiviral signaling by restricting MAVS abundance. It promotes housekeeping degradation of MAVS regardless of the infection signal, through interactions with both MAVS and AIP4. Our findings highlight the inhibitory role for PCBP1 in manipulating both inflammation and innate antiviral immune responses.
Results
PCBP1 inhibits MAVS-mediated activation of IRF3 and NF-κB
To address the potential role of PCBP1 in regulating cellular antiviral signaling, we examined the effects of PCBP1 overexpression on virus-triggered reporter gene activation. Sendai virus (SeV)-induced IFN-β, ISRE and NF-κB activation was inhibited by PCBP1 in a dose-dependent way in 293 cells (Figure 1A), implicating a negative role for PCBP1 in SeV-induced antiviral responses. Next we used both 293 and 293-TLR3 cells (293 cells stably expressing Toll-like receptor 3) to clarify the involvement of PCBP1 in separate pathways, as the synthetic viral double-stranded RNA analog poly(I:C) can only activate IFN-β in TLR3-deficient 293 cells by transfection through the RLR-MAVS pathway; in contrast, 293-TLR3 cells incubated with poly(I:C) show robust IFN-β induction. We found that PCBP1 suppressed transfected but not incubated poly(I:C)-triggered IFN-β activation (Figure 1B), suggesting that it specifically influences intracellular double-stranded RNA signaling. Consistent with that, SeV- and poly(I:C)-triggered secretion of type I IFNs was crippled in cells expressing PCBP1 (Figure 1C). Moreover, PCBP1 dispelled SeV-, Newcastle disease virus (NDV)- and transfected poly(I:C)-induced IRF3 dimerization (Figure 1D), which is a hallmark of IRF3 activation 28. Collectively, these data demonstrate that PCBP1 acts as an inhibitor within the intracellular antiviral pathway.

PCBP1 overexpression inhibits virus-induced signaling. (A, B) Effects of HA-PCBP1 on Luc activation, measured as fold induction relative to empty vector-transfected samples (Vector). Expression of PCBP1 was analyzed by western blot. (A) PCBP1 inhibited IFN-β, ISRE and NF-κB-Luc activation in SeV-infected 293 cells; (B) PCBP1 inhibited IFN-β-Luc activation by transfected poly(I:C) in 293 cells. 293-TLR3, 293 cells stably expressing TLR3; Tran, transfection. (C) PCBP1 depleted type I interferon (IFN) production in SeV-infected and poly(I:C)-transfected 293 cells. N.D., not detected. For (A-C), data are shown as mean ± SD (n = 3). (D) PCBP1 disrupted virus-induced IRF3 dimerization. Native polyacrylamide gel electrophoresis and western blot analyses of lysates from SeV, poly(I:C) and NDV-stimulated 293 cells overexpressing HA-PCBP1 or empty vector. Experiments were repeated three times with similar results.
We coexpressed PCBP1 and components of the cellular antiviral pathway to see if PCBP1 also functions at the MAVS level as PCBP2 does 22. IFN-β induction by MAVS and its upstream RLRs was inhibited by PCBP1, but that by TBK1 and IRF3 was unaffected (Supplementary information, Figure S1A). Measurements of vesicular stomatitis virus (VSV) titers in 293 cells showed that PCBP1 reversed the suppression of viral replication by RIG-I-N (amino acid 1-284), MDA5-N (amino acid 1-206) and MAVS, but not by TBK1 (Supplementary information, Figure S1C). Meanwhile, TRIF-induced reporter activation and IRF3 dimerization were intact (Supplementary information, Figure S1A and S1B). Therefore, PCBP1 selectively inhibits RLR-mediated type I IFN induction at the point of MAVS.
Knockdown of PCBP1 augments cellular antiviral responses
To validate a repression effect for endogenous PCBP1, we used a PCBP1-specific RNAi plasmid 27 to reduce its expression in cells. PCBP1 suppression (Figure 2A) correlated with potentiated IFN-β induction in SeV- and transfected poly(I:C)-stimulated 293 cells (Figure 2B). Consistently, PCBP1 suppression in HeLa cells promoted IFN-β induction by transfected but not incubated poly(I:C) (Figure 2C), indicating its selectivity for the intracellular antiviral pathway. Moreover, PCBP1 depletion prompted an increase in type I IFN production in cells stimulated with SeV or poly(I:C) transfection (Figure 2D). Moreover, PCBP1 knockdown resulted in boosted IFN-β induction triggered by MAVS, RIG-I and MDA5, but not by TRIF, TBK1 or IRF3 (Supplementary information, Figure S1D), and the magnitude of VSV replication declined upon a drop in the level of PCBP1 (Supplementary information, Figure S1E). In summary, these results indicate that PCBP1 negatively regulates MAVS-mediated antiviral signaling.

Knockdown of PCBP1 potentiates cellular antiviral responses. (A) Effect of PCBP1 RNAi versus empty vector and a control RNAi, analyzed by western blot. (B) PCBP1 knockdown potentiated IFN-β-Luc activation by SeV and transfected poly(I:C) in 293 cells. (C) PCBP1 knockdown potentiated IFN-β-Luc activation by poly(I:C) transfection but not by directly adding poly(I:C) to the culture medium of HeLa cells. (D) PCBP1 knockdown increased type I IFN production in SeV-infected and poly(I:C)-transfected 293 cells. N.D., not detected. For (B-D), data are shown as mean ± SD (n = 3).
Interaction of PCBP1 with MAVS
Consistent with a previously identified interaction in the yeast 2-hybrid screen, coimmunoprecipitation experiments in 293 cells revealed that PCBP1 interacted with MAVS, RIG-I and MDA5, but not TRIF or TBK1 (Supplementary information, Figure S2A). In particular, MAVS C-terminus (amino acid 360-540) was both necessary and sufficient for PCBP1 binding, whereas neither the caspase activation and recruitment domain nor the proline-rich region was required (Supplementary information, Figure S2B). Critically, sending MAVS to the endoplasmic reticulum membrane with VAMP-2-TM 3 largely impaired its association with PCBP1, while mitochondria-anchored MAVS chimera containing Bcl-xL-TM could interact normally with PCBP1 (Supplementary information, Figure S2C). These data point to a conclusion that a structural or topological element (or both) unique to the correctly positioned MAVS is important for the interaction.
PCBP1 mediates MAVS degradation via the E3 ubiquitin ligase AIP4
The above results showed functional similarities of PCBP1 and PCBP2 in MAVS inhibition, thus we tested if PCBP1 also mediates MAVS degradation. As we expected, the amount of MAVS protein was much lower upon PCBP1 overexpression while its mRNA remained stable (Figure 3A). Treatment with a proteasome inhibitor MG-132 considerably blocked the protein reduction, while a lysosome inhibitor NH4Cl showed little effect (Figure 3A, upper panel). The finding indicates that PCBP1, similar to PCBP2, causes proteasomal degradation of MAVS. As a further proof, PCBP1 led to Lysine 48 (K48)-linked polyubiquitination of MAVS (Supplementary information, Figure S3A).

PCBP1 mediates MAVS degradation via AIP4. (A) PCBP1 reduced MAVS protein but not mRNA. Top, Western blot analyses of lysates from 293 cells overexpressing Flag-MAVS and increasing amounts of HA-PCBP1 in the presence or absence of MG-132 and NH4Cl; bottom, Relative mRNA levels of MAVS and PCBP1 versus HPRT in untreated samples, determined by reverse transcriptase-PCR. (B) Enhanced MAVS ubiquitination and interaction of MAVS with AIP4 by PCBP1. Coimmunoprecipitation analyses of lysates from 293 cells transfected with the indicated expression plasmids in the presence of MG-132. P1, PCBP1; Ub, ubiquitin. (C) AIP4 aggravated the reduction of MAVS by PCBP1. Western blot analyses of lysates from 293 cells transfected with the indicated plasmids. (D) Loss of MAVS ubiquitination by PCBP1 and AIP4 knockdown. Co-immunoprecipitation analyses of lysates from transfected 293 cells infected with SeV in the presence of MG-132. Experiments were repeated three times with similar results.
Previously, we identified AIP4 as the E3 ubiquitin ligase for MAVS. In fact, it was also recruited by PCBP1 (Supplementary information, Figure S3B) to MAVS-containing complex and enhanced MAVS ubiquitination in the presence of MG-132 (Figure 3B). AIP4 overexpression augmented PCBP1-mediated MAVS degradation (Figure 3C) while its knockdown could minimize SeV-induced MAVS ubiquitination (Figure 3D). Critically, AIP4(C830A), a ligase-deficient version that interacted with PCBP1 (Supplementary information, Figure S3B), failed to synergize with PCBP1 on MAVS degradation (Figure 3C) and even blocked MAVS ubiquitination (Figure 3B). These findings stress the importance of an intact ligase activity of AIP4 during MAVS degradation. On the other hand, sequence alignment revealed that a well-defined AIP4 docking site in PCBP2 is highly conserved in PCBP1 (PASSPV, amino acid 187-192). Mutation of this site [P1(S190A/P191A)] abrogated the association of PCBP1 with AIP4 (Supplementary information, Figure S3B) and hindered AIP4-mediated MAVS ubiquitination (Figure 3B), highlighting the importance of PCBP1 as an adaptor between AIP4 and MAVS. Specifically, loss of AIP4 abrogated the ability of PCBP1 to suppress IFN-β induction by MAVS and RIG-I, but had minimal effect on RNF125 that independently leads to RIG-I and MAVS degradation 29 (Supplementary information, Figure S3C). Collectively, these results suggest that both PCBP1 and PCBP2 facilitate the degradation of MAVS by AIP4.
The synergy of PCBP1 and PCBP2 in regulating MAVS
Now that both PCBPs utilize the same mechanism to regulate MAVS, it is intriguing to find out whether there is a competition or antagonism in between. We investigated the combinatorial actions of PCBP1 and PCBP2 and found that they synergized to inhibit MAVS-induced IFN-β activation in reporter assays, accompanied by enhanced MAVS degradation (Figure 4A). The residual activity of MAVS upon individual PCBP overexpression was almost entirely lost by coexpression of both, reflected by a drop in MAVS-induced type I IFN production (Figure 4B). Furthermore, coexpression of the two proteins resulted in exaggerated suppression on SeV- and poly(I:C)-triggered IFN-β induction (Figure 4C). In all these conditions, PCBP1 and PCBP2 showed a coordinated fashion to inhibit MAVS.

PCBP1 and PCBP2 collaborate to inhibit MAVS. (A, C) Augmented inhibitions of PCBP1 and PCBP2 on IFN-β-Luc activation induced by: (A) MAVS in 293 cells; (C) SeV and transfected poly(I:C) in 293 cells. Protein expression was analyzed by western blot. (B) Enhanced suppression of PCBP1 and PCBP2 on type I IFN production induced by MAVS, but not TRIF in 293 cells. Data are shown as mean ± SD (n = 3).
Different expression patterns of PCBP1 and PCBP2 during viral infection
To address their possible modes of action under physiological conditions, we assessed the distribution of PCBP1 and PCBP2 in mouse organs to see if there was a difference. However, in agreement with a previous report 23, both PCBPs were ubiquitously expressed (Supplementary information, Figure S4A). Then we examined their levels both in resting cells and from stimulated cells. Interestingly, an obvious difference was observed. PCBP1 was stably and abundantly expressed while PCBP2 was robustly induced after stimulation (Figure 5A). Similar results were obtained in SeV-infected A549 (human lung adenocarcinoma epithelial cell line), HT1080 (human fibrosarcoma cell line) and U937 (human leukemic monocyte lymphoma cell line) cells (Figure 5B), indicative of a pervasive expression pattern for the PCBPs in response to viral infection.

Constant expression of PCBP1 and inducible expression of PCBP2. Western blot analyses of lysates from: (A) HeLa cells stimulated with transfected poly(I:C), SeV, NDV and EMCV; (B) A549 cells, HT1080 cells and U937 cells infected with SeV. Experiments were repeated three times with similar results.
Housekeeping degradation of MAVS by PCBP1 in vivo
The expression difference between PCBP1 and PCBP2 is likely related to their physiological functions. To test this, we investigated the effect of each PCBP on MAVS-mediated signaling throughout the infection. First, we confirmed continuous proteasome-dependent degradation of MAVS in both resting and SeV-infected cells, as indicated by its enrichment in the presence of MG-132 (Supplementary information, Figure S4B). Importantly, the level of MAVS increased upon PCBP1 knockdown in untreated cells, but barely changed when PCBP2 was separately silenced (Figure 6A, left). By contrast, MAVS degradation after SeV infection was mostly reversed by PCBP2 knockdown while modestly affected by PCBP1 depletion (Figure 6A, left). Similar effects were observed in SeV-infected HeLa cells (Figure 6A, right). These data confirmed that PCBP2 is a feedback inhibitor while PCBP1 plays a “housekeeping” role. Consistently, the phosphorylation level of IRF3, another hallmark of MAVS activation 30, correlated with the regulatory effects of PCBP1 and PCBP2 after infection (Figure 6A). Further evidence was obtained from both reporter assays (Figure 6B) and VSV plaque assays (Figure 6C), in support of a housekeeping effect for PCBP1 on MAVS-mediated antiviral responses.

Distinct effects of PCBP1 and PCBP2 knockdown on cellular antiviral responses. (A) Western blot analyses of lysates from SeV-infected 293 and HeLa cells transfected separately or simultaneously with PCBP1 and PCBP2 RNAi plasmids. Experiments were repeated three times with similar results. (B) Separate or combined effects of PCBP1 and PCBP2 knockdown on IFN-β-Luc activation by SeV and transfected poly(I:C) in 293 cells. Protein expression was analyzed by western blot. (C) Separate or combined effects of PCBP1 and PCBP2 knockdown on VSV replication in poly(I:C)-transfected 293 cells, determined by standard plaque assays. P1/2, PCBP1/2. For (B) and (C), data are shown as mean ± SD (n = 3).
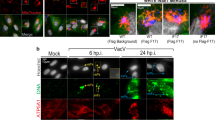
As a prerequisite for regulation, endogenous interaction between PCBP1 and MAVS was detected in 293 cells, regardless of the infection signal (Figure 7A). As an independent means of assessing an in vivo association, HeLa cells were fractionated to separate cytosol, nuclei and mitochondria, which were distinguished by Caspase-3, Histone H3 and COX IV, respectively. A small but constant portion of PCBP1 was traced in the mitochondrial fraction both before and after infection; by contrast, PCBP2 only started to emerge at mitochondria after 4 h of SeV infection (Figure 7B). Double-labeling immunofluorescence studies further confirmed that PCBP1 co-localized with MAVS well before infection (Figure 7C). These data support a preexisting association between endogenous PCBP1 and MAVS, compared to an inducible one for PCBP2 and MAVS. Taken together, we conclude that PCBP1 plays a housekeeping role in vivo to negatively modulate MAVS-mediated antiviral immune responses.

Housekeeping regulations of MAVS by PCBP1. (A) Coimmunoprecipitation analyses of endogenous interactions between PCBP1 and MAVS in SeV-infected 293 cells. IgG, pre-immune mouse serum. (B) Top, fraction diagram used; bottom, Western blot analyses of fractions from SeV-infected HeLa cells. Caspase-3, COX IV and Histone H3 revealed cytosol, mitochondria and nucleus, respectively. For A and B, experiments were repeated three times with similar results. (C) Cells in B were subject to immunofluorescence microscopy, showing the locations of endogenous PCBP1 (green) and MAVS (red). Nuclei were stained with DAPI. Images were representative of three independent experiments. (D) A schema showing housekeeping restriction of MAVS by PCBP1. Please see the text for details.
Discussion
RIG-I-like receptors play a major role in sensing RNA virus infection and enlist the adaptor MAVS to initiate and modulate antiviral immunity 31. MAVS is reported to reside on the outer membranes of the mitochondria and peroxisomes 8, 32 where it signals rapid induction of antiviral cytokines including type I IFNs. A tight MAVS regulation is essential to prevent unwarranted responses or even inflammation 33. The regulation occurs at various levels ranging from complex formation to posttranslational modifications 34, among which ubiquitination and degradation by the proteasome pathway have been frequently reported 22, 29, 35, 36.
Following the study on PCBP2 22, we here characterized PCBP1 as another negative regulator in cellular antiviral signaling through MAVS degradation. The expression patterns of PCBP1 and PCBP2 support the regulation model we proposed (Figure 7D): at the steady phase, MAVS is constantly restricted by PCBP1 to prevent spontaneous activation of the immune responses. Upon viral infection, rapidly boosted signaling turns in favor of positive signal amplification and numerous IFN-stimulated genes (including PCBP2) are induced (the active phase). After that, PCBP2 serves as part of the negative feedback loop to shut down MAVS signaling (the terminal phase).
As expected, PCBP1 and PCBP2 adopt the same mechanism to promote MAVS degradation (Figure 3). This result conforms to our previous report that the “WB2” motif of PCBP2 is a prerequisite for AIP4 binding 22, since a highly conserved motif in PCBP1 is also indispensible for AIP4 recruitment (Figure 3C and Supplementary information, Figure S3B). As such, it appears that the homology in sequence could not be fortuitous, as the PCBP1 locus was reported as the retrotransposition product of a minor PCBP2 splicing variant 37. Also, the synergy of PCBP1 and PCBP2 we observed (Figure 4) is consistent with their complementary functions reported before 25, 26, 27, 38, 39.
The present study demonstrates that PCBP1 holds a distinct role in vivo compared to PCBP2 in regulating cellular antiviral signaling, reflected by their different expression patterns, including: (1) the basal level of PCBP1 is much higher than that of PCBP2; and (2) viral infection triggers a sharp increase in PCBP2 abundance without affecting the overall amount of PCBP1 (Figure 5). The unique property of PCBP1 was observed in knockdown studies that point to a housekeeping role for PCBP1 at the quiescent state (Figure 6). This specificity may serve as another sound piece of evidence for divergent physiological functions of the two proteins as reported elsewhere 40, 41, 42, 43.
Our investigation extends the multiple capacity of PCBP1 43, 44 as an E3 ubiquitin ligase adaptor for housekeeping degradation of MAVS 45, 46. Besides, PCBP1 displays some regulatory effects after viral infection (Figures 3D and 6). In either case, the engagement of PCBP1 is crucial because of one thing, effective suppression of IFN-α/β may only be possible with the concerted actions of several suppressors, and for the other, unlike those stimulus-triggered inhibitors including PCBP2 6, 22, 35, 47, 48, 49, 50, 51, 52, 53, PCBP1 may serve to tune the threshold for initiation of signal transduction by restricting MAVS at basal conditions 54, 55. The latter feature of PCBP1 is of special importance in preventing various inflammatory disorders caused by unwanted IFN 33, 56, 57, 58 as a result of possible activities of excessive MAVS. On the other side, MAVS degradation by PCBP2 in those infected cells may seem rather counterproductive for self-protection. Fortunately, PCBP2 belongs to the post-infection inhibitors whose expression is only induced by an infection signal. That said, PCBP2 is expressed at such a low level before stimulation that it can hardly hamper the initiation of proper antiviral responses, but instead functions as a delayed regulator upon induction.
In conclusion, our studies uncover an important role for PCBP1 in immune modulation through housekeeping degradation of MAVS. The finding that both PCBPs punctually fine-tune the activity of MAVS not only highlights the pleiotropic effects of poly(C)-binding proteins, but also implicates an evolutionarily non-redundant function for PCBP1 compared to PCBP2.
Materials and Methods
Plasmids
Mammalian expression plasmids for Flag- and HA-tagged PCBP1, HA-RNF125 and Myc-ubiquitin (Ub) were constructed by standard molecular biology techniques. PCBP1(S190A/P191A) was generated using the QuickChange Site-directed Mutagenesis Kit (Stratagene) with the following primers: 5′-G CCC ATG CCG GCC AGC GCC GCA GTC ATC TGC GCG GGC G-3′ and 5′-C GCC CGC GCA GAT GAC TGC GGC GCT GGC CGG CAT GGG C-3′. All constructs were verified by sequencing. HA-Ub(K48) and HA-Ub(K63) were kindly provided by Dr Hong-Bing Shu (Wuhan University, Wuhan, China). Flag-tagged MAVS (VAMP-2-TM) and MAVS (Bcl-xL-TM) were kind gifts from Dr Zhijian J Chen (University of Texas Southwestern Medical Center, Dallas, USA). Other plasmids were previously described 22.
Reagents
MG-132 was purchased from Calbiochem. PCBP1 antiserum was generated by immunizing mice with a recombinant His6-PCBP1 protein produced in E. coli, at Animal Facility, Institute of Genetics & Developmental Biology, Chinese Academy of Sciences, China. Viruses and other reagents were previously described 22, 59.
Cell culture, transfection, viral infection and treatment
U937 cells were grown in a 5% CO2 humidified atmosphere in RPMI1640 (Gibco) with penicillin-streptomycin (Gibco) and 10% fetal bovine serum (Hyclone). Other cells were maintained in Dulbecco's modified Eagle medium (Gibco) with 10% fetal bovine serum and antibiotics. Transient transfection was carried out by the standard calcium phosphate precipitation method. Poly(I:C) (1 μg/ml) was transfected using GenEscort II (Wisegen) according to the manufacturer's instructions. Where indicated, cells were infected with SeV (multiplicity of infection (MOI, 1), VSV (MOI, 0.1), NDV-GFP (MOI, 8) or EMCV (MOI, 5) in serum-free medium. One hour later, cells were rinsed and cultured in fresh medium for the indicated times. Cycloheximide (100 mg/ml), poly(I:C) (10 μg/ml), MG-132 (20 mM) and NH4Cl (20 mM) were applied to culture medium at the indicated concentrations.
Luciferase (Luc) reporter assay
Cells (1 × 105) were transfected with 100 ng of a Luc reporter gene, 50 ng of pRL-SV40-Renilla (Promega) as an internal control and the indicated expression vectors for 24 h. Reporter gene activity was analyzed using the Dual-Luciferase Reporter 1000 Assay System (Promega) and measured with a TD-20/20 Luminometer (Turner Designs) according to the manufacturers' instructions. Three independent experiments were carried out in duplicate. Error bars represent the mean standard deviations (mean ± SD).
Protein extraction, immunoprecipitation, western blot and Native polyacrylamide gel electrophoresis
Cells were lysed in cold lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 2 mM ethylene glycol tetraacetic acid, 10 mM NaF, 1 mM Na3VO4 and 2 mM dithiothreitol) containing protease inhibitors. Immunoprecipitation was carried out in lysates with 20 μl of Protein A Sepharose (Amersham Biosciences) and 1 μg of the indicated antibody at 4 °C for 4 to 12 h with constant agitation. Following three washes in cold lysis buffer or phosphate-buffered saline, samples were eluted with 2 × SDS sample buffer by boiling at 99 °C for 10 min. Western blot was carried out by standard procedures; proteins were visualized with an Odyssey Two-Color Infrared Imaging System (LI-COR Biosciences, USA) according to the manufacturer's instructions.
Native gel electrophoresis and IRF3 dimerization assays were carried out as described previously 22. Briefly, lysates were mixed with 2× Native sample buffer (0.125 mM Tris-HCl, pH 6.8, 30% glycerol and 2% deoxycholate) before loading and electrophoresis at 25 mA for 1 h on ice.
Type I IFN bioassay
Type I IFN activity was measured as previously described 22.
RNA interference
Double-stranded oligonucleotides targeting human PCBP1 (5′-GGG AGA GTC ATG ACC ATT C-3′ 27) were cloned into the pSuper.Retro RNAi plasmid (Oligoengine Inc.). PCBP2 and AIP4 RNAi plasmids were previously described 22. For silencing experiments, cells (5 × 105) were transfected with 3 μg of RNAi plasmids for 48 h prior to further treatment.
VSV plaque assay
293 cells (1 × 105) were transfected with the indicated expression plasmids for 24 h before infection with VSV in serum-free medium for 1 h. Cells were rinsed and incubated in fresh medium for 23 h. Proper dilutions of the medium containing VSV were inoculated onto confluent monolayer of BHK21 cells for 1 h before fresh medium containing 2% methylcellulose was added for 60 h. Cells were fixed in 4% formaldehyde and stained with 0.2% crystal violet dissolved in 20% methanol. Plaques were counted, averaged and multiplied by the dilution rates for VSV titers measured in plaque forming units per ml.
Immunofluorescence microscopy
Cells on coverslips were fixed in cold methanol for 15 min. After three washes in phosphate buffered saline, cells were blocked in phosphate buffered saline containing 5% bovine serum albumin for 30 min, and incubated with mouse α-PCBP1 and rabbit α-MAVS primary antibodies in phosphate buffered saline containing 3% bovine serum albumin for 2 h at 37 °C. After three washes, cells were incubated with FITC (fluorescein isothiocyanate)-conjugated α-mouse and TXRD-conjugated α-rabbit secondary antibodies (Imgenex) for 1 h at 37 °C and then with DAPI (4′,6-Diamidino-2-phenylindole, Roche) for 15 min. The coverslips were washed extensively and mounted onto slides. Imaging of the cells was carried out using an Olympus BX51 microscope under a ×100 oil objective.
RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen). After reverse transcription with oligo(dT) primer using a RevertAid First Strand cDNA Synthesis Kit (Fermentas), aliquots of products were subject to standard PCR. Gene-specific primer sequences are as follows: PCBP1, 5′-GGA TGC CGG TGT GAC TGA AAG-3′ and 5′-TTA CAC CCG CCT TTC CCA ATC-3′; MAVS, 5′-CCG TTT GCT GAA GAC AAG-3′ and 5′-CTA GTA GCT CTG GTA GAC AGA GGC-3′; HPRT, 5′-CTG GCG TCG TGA TTA GTG ATG-3′ and 5′-TAT CCA ACA CTT CGT GGG GTC-3′.
Subcellular fractionation
HeLa cells were rinsed and lysed in cold homogenization buffer (buffer H: 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 0.2 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol and protease inhibitors). The homogenate (S0) was centrifuged at 500× g for 10 min and the pellet was resuspended in buffer H as crude nuclei (P1). The supernatant was centrifuged at 5 000× g for 10 min to precipitate crude mitochondria (P5).
Statistics
The student's t-test was used for a comparison of two independent treatments. For all tests, a P value < 0.05 was considered significant (*) while P < 0.01, very significant (**).
References
Yoneyama M, Fujita T . RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev 2009; 227:54–65.
Wilkins C, Gale M Jr . Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol 2010; 22:41–47.
Seth RB, Sun L, Ea CK, Chen ZJ . Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005; 122:669–682.
Xu LG, Wang YY, Han KJ, et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 2005; 19:727–740.
Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005; 6:981–988.
Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437:1167–1172.
Scott I . The role of mitochondria in the mammalian antiviral defense system. Mitochondrion 2010; 10:316–320.
Dixit E, Boulant S, Zhang Y, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010; 141:668–681.
Clement JF, Meloche S, Servant MJ . The IKK-related kinases: from innate immunity to oncogenesis. Cell Res 2008; 18:889–899.
Nakhaei P, Genin P, Civas A, Hiscott J . RIG-I-like receptors: sensing and responding to RNA virus infection. Semin Immunol 2009; 21:215–222.
Yoneyama M, Fujita T . Recognition of viral nucleic acids in innate immunity. Rev Med Virol 2010; 20:4–22.
Takeuchi O, Akira S . Innate immunity to virus infection. Immunol Rev 2009; 227:75–86.
Seth RB, Sun L, Chen ZJ . Antiviral innate immunity pathways. Cell Res 2006; 16:141–147.
Sadler AJ, Williams BR . Interferon-inducible antiviral effectors. Nat Rev Immunol 2008; 8:559–568.
Kumar H, Kawai T, Kato H, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med 2006; 203:1795–1803.
Sun Q, Sun L, Liu HH, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 2006; 24:633–642.
Lin R, Paz S, Hiscott J . Tom70 imports antiviral immunity to the mitochondria. Cell Res 2010; 20:971–973.
Ting JP, Duncan JA, Lei Y . How the noninflammasome NLRs function in the innate immune system. Science 2010; 327:286–290.
Richards KH, Macdonald A . Putting the brakes on the anti-viral response: negative regulators of type I interferon (IFN) production. Microbes Infect 2011; 13:291–302.
Komuro A, Bamming D, Horvath CM . Negative regulation of cytoplasmic RNA-mediated antiviral signaling. Cytokine 2008; 43:350–358.
Liu SY, Sanchez DJ, Cheng G . New developments in the induction and antiviral effectors of type I interferon. Curr Opin Immunol 2011; 23:57–64.
You F, Sun H, Zhou X, et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol 2009; 10:1300–1308.
Leffers H, Dejgaard K, Celis JE . Characterisation of two major cellular poly(rC)-binding human proteins, each containing three K-homologous (KH) domains. Eur J Biochem 1995; 230:447–453.
Kiledjian M, Wang X, Liebhaber SA . Identification of two KH domain proteins in the alpha-globin mRNP stability complex. EMBO J 1995; 14:4357–4364.
Evans JR, Mitchell SA, Spriggs KA, et al. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene 2003; 22:8012–8020.
Kim SS, Pandey KK, Choi HS, et al. Poly(C) binding protein family is a transcription factor in mu-opioid receptor gene expression. Mol Pharmacol 2005; 68:729–736.
Waggoner SA, Johannes GJ, Liebhaber SA . Depletion of the poly(C)-binding proteins alphaCP1 and alphaCP2 from K562 cells leads to p53-independent induction of cyclin-dependent kinase inhibitor (CDKN1A) and G1 arrest. J Biol Chem 2009; 284:9039–9049.
Servant MJ, Grandvaux N, Hiscott J . Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem Pharmacol 2002; 64:985–992.
Arimoto K, Takahashi H, Hishiki T, et al. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA 2007; 104:7500–7505.
Servant MJ, Grandvaux N, tenOever BR, et al. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J Biol Chem 2003; 278:9441–9447.
Loo YM, Gale M Jr . Immune signaling by RIG-I-like receptors. Immunity 2011; 34:680–692.
Sharma S, Fitzgerald KA . Viral defense: it takes two MAVS to Tango. Cell 2010; 141:570–572.
Takeuchi O, Akira S . Pattern recognition receptors and inflammation. Cell 2010; 140:805–820.
Moore CB, Ting JP . Regulation of mitochondrial antiviral signaling pathways. Immunity 2008; 28:735–739.
Jia Y, Song T, Wei C, et al. Negative regulation of MAVS-mediated innate immune response by PSMA7. J Immunol 2009; 183:4241–4248.
Zhong B, Zhang L, Lei C, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 2009; 30:397–407.
Makeyev AV, Chkheidze AN, Liebhaber SA . A set of highly conserved RNA-binding proteins, alphaCP-1 and alphaCP-2, implicated in mRNA stabilization, are coexpressed from an intronless gene and its intron-containing paralog. J Biol Chem 1999; 274:24849–24857.
Ostareck-Lederer A, Ostareck DH, Hentze MW . Cytoplasmic regulatory functions of the KH-domain proteins hnRNPs K and E1/E2. Trends Biochem Sci 1998; 23:409–411.
Makeyev AV, Liebhaber SA . The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA 2002; 8:265–278.
Zhu Y, Sun Y, Mao XO, Jin KL, Greenberg DA . Expression of poly(C)-binding proteins is differentially regulated by hypoxia and ischemia in cortical neurons. Neuroscience 2002; 110:191–198.
Woolaway K, Asai K, Emili A, Cochrane A . hnRNP E1 and E2 have distinct roles in modulating HIV-1 gene expression. Retrovirology 2007; 4:28.
Sean P, Nguyen JH, Semler BL . The linker domain of poly(rC) binding protein 2 is a major determinant in poliovirus cap-independent translation. Virology 2008; 378:243–253.
Chaudhury A, Chander P, Howe PH . Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: focus on hnRNP E1's multifunctional regulatory roles. RNA 2010; 16:1449–1462.
Huo LR, Shen C, Ju WN, et al. Identification of novel partner proteins of PCBP1. Beijing Da Xue Xue Bao 2009; 41:402–408.
Meng Q, Rayala SK, Gururaj AE, et al. Signaling-dependent and coordinated regulation of transcription, splicing, and translation resides in a single coregulator, PCBP1. Proc Natl Acad Sci USA 2007; 104:5866–5871.
Shi H, Bencze KZ, Stemmler TL, Philpott CC . A cytosolic iron chaperone that delivers iron to ferritin. Science 2008; 320:1207–1210.
Li XD, Sun L, Seth RB, Pineda G, Chen ZJ . Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA 2005; 102:17717–17722.
Lin R, Lacoste J, Nakhaei P, et al. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J Virol 2006; 80:6072–6083.
Loo YM, Owen DM, Li K, et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci USA 2006; 103:6001–6006.
Cheng G, Zhong J, Chisari FV . Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc Natl Acad Sci USA 2006; 103:8499–8504.
Chen Z, Benureau Y, Rijnbrand R, et al. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J Virol 2007; 81:964–976.
Yang Y, Liang Y, Qu L, et al. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci USA 2007; 104:7253–7258.
Zhong B, Zhang Y, Tan B, et al. The E3 ubiquitin ligase RNF5 targets virus-induced signaling adaptor for ubiquitination and degradation. J Immunol 2010; 184:6249–6255.
Moore CB, Bergstralh DT, Duncan JA, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008; 451:573–577.
Yasukawa K, Oshiumi H, Takeda M, et al. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal 2009; 2:ra47.
Pascual V, Chaussabel D, Banchereau J . A genomic approach to human autoimmune diseases. Annu Rev Immunol 2010; 28:535–571.
Trinchieri G . Type I interferon: friend or foe? J Exp Med 2010; 207:2053–2063.
Theofilopoulos AN, Baccala R, Beutler B, Kono DH . Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 2005; 23:307–336.
Sun W, Li Y, Chen L, et al. An endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA 2009; 106:8653–8658.
Acknowledgements
We thank Drs H Shu (Wuhan University) for HA-Ub(K48) and HA-Ub(K63) plasmids, and Z Chen (University of Texas Southwestern Medical Center) for Flag-MAVS (VAMP-2-TM) and Flag-MAVS (Bcl-xL-TM) plasmids. We are grateful to members of the Jiang laboratory for discussion. This work is supported by grants from the China Natural Science Foundation (numbers 30772024 and 31025010), the Chinese Ministry of Science and Technology (number 2007CB914502) and the Chinese Ministry of Education (number 108002).
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
PCBP1 inhibits antiviral signaling at the level of MAVS. (PDF 134 kb)
Supplementary information, Figure S2
PCBP1 interacts with MAVS. (PDF 231 kb)
Supplementary information, Figure S3
AIP4 is responsible for PCBP1–mediated MAVS degradation. (PDF 100 kb)
Supplementary information, Figure S4
Expression of PCBPs and MAVS. (PDF 50 kb)
Rights and permissions
About this article
Cite this article
Zhou, X., You, F., Chen, H. et al. Poly(C)-binding protein 1 (PCBP1) mediates housekeeping degradation of mitochondrial antiviral signaling (MAVS). Cell Res 22, 717–727 (2012). https://doi.org/10.1038/cr.2011.184
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2011.184
Keywords
This article is cited by
-
Molecular and functional characterization of porcine poly C binding protein 1 (PCBP1)
BMC Veterinary Research (2024)
-
PCBP1 modulates the innate immune response by facilitating the binding of cGAS to DNA
Cellular & Molecular Immunology (2021)
-
Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors
Cellular & Molecular Immunology (2021)
-
Regulation of antiviral innate immune signaling and viral evasion following viral genome sensing
Experimental & Molecular Medicine (2021)
-
RNF115 plays dual roles in innate antiviral responses by catalyzing distinct ubiquitination of MAVS and MITA
Nature Communications (2020)
