
Abstract
Caspase-2 represents the most conserved member of the caspase family, which exhibits features of both initiator and effector caspases. Using ribonucleoprotein (RNP)-immunoprecipitation assay, we identified the proapoptotic caspase-2L encoding mRNA as a novel target of the ubiquitous RNA-binding protein HuR in DLD-1 colon carcinoma cells. Unexpectedly, crosslinking-RNP and RNA probe pull-down experiments revealed that HuR binds exclusively to the caspase-2-5′ untranslated region (UTR) despite that the 3′ UTR of the mRNA bears several adenylate- and uridylate-rich elements representing the prototypical HuR binding sites. By using RNAi-mediated loss-of-function approach, we observed that HuR regulates the mRNA and in turn the protein levels of caspase-2 in a negative manner. Silencing of HuR did not affect the stability of caspase-2 mRNA but resulted in an increased redistribution of caspase-2 transcripts from RNP particles to translational active polysomes implicating that HuR exerts a direct repressive effect on caspase-2 translation. Consistently, in vitro translation of a luciferase reporter gene under the control of an upstream caspase-2-5′UTR was strongly impaired after the addition of recombinant HuR, whereas translation of caspase-2 coding region without the 5′UTR is not affected by HuR confirming the functional role of the caspase-2-5′UTR. Functionally, an elevation in caspase-2 level by HuR knockdown correlated with an increased sensitivity of cells to apoptosis induced by staurosporine- and pore-forming toxins as implicated by their significant accumulation in the sub G1 phase and an increase in caspase-2, -3 and poly ADP-ribose polymerase cleavage, respectively. Importantly, HuR knockdown cells remained insensitive toward STS-induced apoptosis if cells were additionally transfected with caspase-2-specific siRNAs. Collectively, our findings support the hypothesis that HuR by acting as an endogenous inhibitor of caspase-2-driven apoptosis may essentially contribute to the antiapoptotic program of adenocarcinoma cells by HuR.
Similar content being viewed by others

Main
An important feature of apoptotic cell death is the activation of caspases, a family of cysteine-aspartate proteases, which mediate the proteolytic degradation of diverse downstream substrates (for recent reviews, see Kumar;1 Riedl and Shi;2 Bouchier-Hayes3). Caspases are divided into two main classes, the initiator caspases including caspase-1, -8, -9 and -10 and the effector caspases-3, -6 and -7.4, 5 Strikingly, the role of caspase-2, the evolutionarily most conserved caspase, in regulating apoptosis remains obscure (for a review, see Kitevska et al.5). Although the cleavage specificity of caspase-2 resembles that of effector caspases, caspase-2 shares various features of an initiator caspase as it contains a long caspase activation and recruitment domain, which is necessary for dimerization and activation.6 Mechanistically, caspase-2 was shown to be recruited to the multimeric PIDDosome protein complex for its activation during DNA damage-mediated apoptosis.6 In addition, a broad range of external stimuli can also activate caspase-2 through a typical ‘induced-proximity’ mechanism, which includes a dimerization of the protein, thereby allowing for autoprocessing of the inactive zymogens.7, 8, 9 Intriguingly, an initiator caspase role for caspase-2 could be furthermore demonstrated for apoptosis of epithelial cells induced by pore-forming bacterial toxins independent of PIDDosome formation.10, 11 Genetically, the proapoptotic and tumor-suppressive activity is specifically confined to the long isoform of caspase-2 (caspase-2L also called ICH-1L) derived from alternative splicing of caspase-2 encoding mRNA, which is expressed in a broad range of tissues12, 13, 14. Conversely, overexpression of the short caspase-2 isoform, the caspase-2S (ICH-1S) inhibits apoptosis but importantly, expression of this splice variant was only demonstrated in some tissues.12, 15
The reduced sensitivity towards apoptotic signals is a hallmark of tumorigenesis, which is achieved by the enhanced activation of oncogenic cell survival pathways in the tumor cells. Thereby, emerging experimental evidence of the last decade implicates that besides variations in the transcriptome of tumor cells, posttranscriptional mechanisms have a crucial role for an increased cell survival of tumor cells (for a previous review, see Abdelmohsen et al.16). The ubiquitous RNA-binding protein Human antigen R (HuR) is increasingly recognized as a key player in the deregulated posttranscriptional control of many oncogenes. It was shown by many publications that HuR can protect cells from apoptotic cell death either by stabilizing and/or enhancing the translation of target mRNAs coding for prosurvival factors or by inhibiting the translation of proapoptotic proteins. Likewise, enhanced HuR expression was observed in many human tumors17, 18, 19, 20, 21 and increased levels of total and/or cytoplasmic HuR correlate with a high grade malignancy as convincingly demonstrated for example, in human colorectal cancer.22 Mechanistically, HuR stabilizes its target mRNA mainly through specifically binding to adenylate- and uridylate-rich elements (AREs) usually located in the 3′untranslated region (UTR) of a large subset of labile mRNAs. As mentioned, in addition to acting as an mRNA stability factor, HuR can also bind mRNAs and thereby directly affect their translation23, 24, 25, 26, 27 or, alternatively, trigger micro-RNA-mediated gene repression (for a previous review see Srikantan et al.28).
In this study, by using a ribonucleoprotein-immunoprecipitation (RNP-IP)-Chip analysis, we have identified caspase-2L as a novel target gene of HuR and interestingly, HuR constitutively bound to the 5′UTR, but not to the 3′UTR of the mRNA. With regard to the high HuR abundance observed in most cancers and tumor cells, we used an siRNA approach to determine the functional consequences of HuR depletion. Interestingly, siRNA knockdown of HuR caused a significant elevation in caspase-2L levels and is concomitant with a markedly increased sensitivity of adenocarcinoma cells toward STS- or α-toxin-induced apoptosis. The negative regulation of caspase-2L by HuR could represent a novel mechanism of the general antiapoptotic program by HuR and thereby provides an additional explanation for the increased resistance of adenocarcinoma cells towards extracellular death signals.
Results
Identification of Caspase-2 mRNA as a novel candidate HuR target in DLD-1 cells
By using RIP-Chip assay in the human colon carcinoma cell line DLD-1, we identified a total number of 307 mRNAs, which were enriched more than 1.5-fold in the HuR-RIP-Chip if compared with the IgG control group (Supplementary Table). Analyzing RIP-Chip data by Ingenuity software revealed that among these HuR-enriched mRNAs 49 mRNA species can be functionally assigned to cell death and survival (marked rows of Supplementary Table). For further investigations, we focused on caspase-2 as the defined role of this evolutionally most conserved caspase in apoptotic pathways is not fully understood.5 Furthermore, the 3′UTR of the caspase-2-encoding mRNA contains several prototypical HuR binding sites including several pentameric AREs and U-rich elements, respectively. Indeed, a strong binding of HuR to caspase-2 mRNA in DLD-1 cells was confirmed by pull-down RT-PCR assay (Figure 1a, upper panel). Similarly, cyclooxygenase-2 (Cox-2) and cyclin A mRNAs, two well-established HuR targets in colon carcinoma cells,22, 29 were highly enriched in the HuR precipitates (Figure 1a, left panel). Western blot analysis furthermore confirmed the specific binding of the antibody used for pull-down RT-PCR (Figure 1a, right panel).

Constitutive binding of HuR to the 5′UTR of Caspase-2 mRNA in DLD-1 cells. (a) An RNP-IP assay was performed in combination with RT-PCR from total cell lysates of DLD-1 cells to amplify mRNA species specifically bound by HuR. HuR-bound mRNA was precipitated by addition of a monoclonal HuR antibody (HuR) or, alternatively, the same amount of mouse IgG (IgG) as negative control. After elution from Sepharose beads, the isolated RNA samples were analyzed by RT-PCR using primer pairs, complementary and specific to the coding region of the indicated genes. The length of corresponding PCR products was determined by a DNA ladder. The specific immunoprecipitation (IP) of HuR is confirmed by western blot analysis (W.blot) shown in the right part of the panel. Data shown are representative of two independent experiments giving similar results. (b, upper panel) Schematic representation of the different splice variants from human caspase-2 mRNA including caspase-2L (v1), caspase-2S (v2), and variant 3 (v3) encoding transcripts. The relative position of start (gray) and stop (dark) codons is depicted by triangles. Boxes show the relative position of exons and lacking exons are symbolized by a continuous line. The doubleheaded arrows indicate different regions (I, III and IV) encompassing variant-specific sequences (lines) or a region (II) commonly found in all three splice variants (dotted). (b, lower panel) Crosslinking-RNP-IP analysis demonstrating that HuR specifically interacts with the 5′UTR of caspase-2L (v1) and splice variant 3, respectively. RNA-protein interactions were crosslinked with formaldehyde before HuR-bound mRNA was immunoprecipitated by addition of a HuR-specific antibody (HuR) or, alternatively, by the same amount of mouse IgG (IgG). The precipitated RNA samples were analyzed by RT-PCR by using primer pairs, spanning the indicated regions of caspase-2 mRNA (roman numerals). Input levels of different caspase-2 transcripts were isolated before the IP and were assessed by RT-PCR using the same primer pairs. (c) Biotin pull-down assay demonstrating a specific HuR binding to the 5′UTR of caspase-2. For pull-down assay, the biotinylated transcript encompassing 241 nucleotides of the 5′UTR of human caspase-2 (5′UTR Casp-2) was incubated with 300 μg of total cell lysates from DLD-1 cells. The specific binding of HuR in the pull-down material was confirmed by western blotting. Biotinylated RNA from partial human reverse GAPDH (hrg) was used as a negative control, whereas 0.1 μg of recombinant HuR (GST-HuR) instead of cell lysates was incubated with the same amount of biotinylated 5′UTR of caspase-2 and used as a positive control. Data shown are representative of two independent experiments giving similar results
Constitutive binding of HuR to the 5′UTR of caspase-2 mRNA in DLD-1 cells
The human caspase-2 gene (ICH-1) is target of alternative splicing, which results in the generation of either an exon 9 lacking proapoptotic caspase-2L transcript (variant 1 or synonym: ICH-1L or Nedd-2) expressed in a wide range of different tissues or, alternatively, an exon 9 containing and antiapoptotic caspase-2S transcript (variant 2, synonymously called ICH-1S), which was only detected in some tissues. In contrast, the functional impact of the third splice variant (variant 3) with truncated exons is unknown.3, 13 A common structural feature of these three splice variants is the presence of several typical HuR binding sites (Figure 1b, upper panel). To test whether HuR would preferentially bind to only a specific caspase-2 transcript, we performed a crosslinking-RNP-IP assay by using specific primer pairs encompassing specific regions of the different splice variants (roman numerals in Figure 1b). Before, the specificity of HuR binding was confirmed by the inability of control IgG to yield appropriate PCR products (‘IgG’). HuR binds to a region that is found in the common 5′UTR of v1 and v3 (‘5′UTR Casp-2-v1,v3’), whereas no amplificated cDNA was visible when a primer pair encompassing a region present in the 3′UTR of all three mentioned caspase-2 splice variants was used. Also, no amplification product was detectable with primer pairs encompassing a region that is only found in the 3′UTR of caspase-2S (‘3′UTR Casp-2-v2’) or with primer pairs covering a region that is exclusively found in the 3′UTR of splice variant 3 (‘3′UTR Casp-2-v3’). Interestingly, control PCRs from the input material (Input) showed the presence of all three caspase-2 splicing variants (v1, v2, v3) in DLD-1 cells (Figure 1b, lower panel). We additionally performed streptavidin-tethered RNA-affinity purification using in vitro transcribed and biotin-labeled 5′UTR of caspase-2L. Thereby, we detected a specific binding of HuR to caspase-2L-5′UTR, whereas no immunopositive signal was observed when using a control RNA of similar length encoding partial human glyceraldehyde-3 phosphate dehydrogenase (GAPDH) in reverse orientation (‘hrg’) (Figure 1c). These results confirm the constitutive HuR binding to the 5′UTR of the mRNA coding for the proapoptotic caspase-2L.
HuR knockdown by siRNA increases the steady-state levels of caspase-2 in human colon carcinoma cells without affecting its mRNA stability
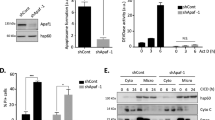
Next, we tested whether HuR binding to caspase-2L has a functional impact on caspase-2L expression by analyzing whether depletion of HuR by small interfering (si) RNA would influence the mRNA levels of caspase-2L. Administering a mixture of four different HuR-specific siRNAs for 48 h resulted in a robust decrease of almost 80% in HuR-mRNA levels when compared with DLD-1 cells transfected with a control siRNA (siCtrl.) (Figure 2a). Although untypical but in accordance to our finding, that HuR did not bind to the typical AREs present in the 3′UTR of the three different caspase-2 splice variants, the steady-state level of caspase-2L mRNA were significantly elevated in HuR-siRNA-depleted cells when compared with control-siRNA transfectants (Figure 2a). Monitoring caspase-2L mRNA decay with the transcriptional inhibitor actinomycin D revealed that the stability of caspase-2L mRNA was not influenced by the siRNA-mediated knockdown of HuR. In contrast, the levels of Cox-2 mRNA, a well-known target of HuR-dependent mRNA stabilization, were clearly decreased in HuR-silenced DLD-1 cells (Figure 2b).

HuR knockdown by siRNA increases the steady-state levels of caspase-2 mRNA in human colon carcinoma cells without affecting its mRNA stability. (a) DLD-1 cells were transfected with either siRNA duplexes of HuR (siHuR), or control-siRNA duplexes (siCtrl.) as described in the Materials and Methods section. Forty-eight hours after transfection, cells were harvested and extracted for total cellular RNA and changes in HuR and caspase-2 mRNA levels were determined by quantitative reverse transcription-(qRT)-PCR using β-actin mRNA for normalization. The values represent means±S.D. (n=3) and are depicted as -fold induction (P***≤0.001) compared with the levels of corresponding mRNA in control-siRNA transfectants. (b) Schematic representation of caspase-2L (v1) mRNA. The relative positions of U-rich elements (gray ovals) and pentameric AUUUA sequences (open boxes) in the 3′UTR of the mRNA and the length of the coding region are indicated by a kilo base (kb)-scale. DLD-1 cells were transfected with siRNA duplexes of HuR (siHuR) or control-siRNA duplexes (siCtrl.) for 48 h before transcription was blocked by the addition of actinomycin D (5μg/ml). After the indicated time periods, cells were harvested and extracted for total cellular RNA. Graphs show means±S.D. (n=3) and depict the percentage of remaining caspase-2L (left panel) or Cox-2 (right panel) mRNA levels in relation to β-actin mRNA and compared with the levels of normalized mRNA species measured immediately before the addition of actinomycin D and that were set as 100%
A similar effect by HuR silencing was observed on the protein level of caspase-2L (Figure 3a, left panel). Importantly, an inverse correlation of both proteins was also observed in SW620 cells, thus excluding a cell line-specific phenomenon (Figure 3a, right panel). In contrast to caspase-2, the levels of cyclin A which represents another well-known target of HuR-dependent mRNA stabilization was strongly reduced after the knockdown of HuR, whereas the level of β-tubulin and GAPDH remained unchanged (Supplementary Figure S1). Monitoring the time-course of caspase-2L decay in response to cycloheximide by western blot analysis furthermore indicates that the increase in caspase-2L protein in HuR-knockdown cells does not rely on an elevated protein stability (Figure 3b, left panel, Supplementary Figure S2). Together with our observations that HuR does constitutively bind to the 5′UTR of caspase-2, these data indicate that translation of caspase-2L is suppressed by HuR. To test whether siRNA knockdown of HuR would affect the translation status of DLD-1 cells, total lysates from cells were layered on a sucrose cushion for polysomal fractionation.30 Owing to the limited yield of input material from subconfluent transfectants, we chose a protocol which crudely separates fractions collecting low- and high-molecular weight polysomes from nonpolysomal fractions containing translational inactive RNPs.31 The RNA from both pools was extracted and the relative distribution of caspase-2L mRNA in both transfectants was quantified by semiquantitative RT-PCR. We observed a clear increase in the relative polysome-to-RNP ratio of caspase-2 mRNA, indicating a switch of caspase-2 mRNA distribution from nonpolysomal (RNPs) to the polysomal fraction under conditions where HuR expression is diminished (Figure 3b, right panel).

HuR inhibits translation of caspase-2L via binding to its 5′UTR. (a) DLD-1 (left panel) or SW620 (right panel) colonic carcinoma cells were either transfected with duplex siRNA of HuR (siHuR) or with control siRNA (siCtrl.). Forty-eight hours after transfection, cells were lysed and total protein (30 μg) subjected to SDS-PAGE and successively immunoblotted with anti-HuR, anti-caspase-2 and anti-β-actin antibodies, of which the latter one was used as a loading control. The data shown are representative of three independent experiments giving similar results. (b, left panel) Subconfluent DLD-1 cells were transfected with siRNA duplexes of HuR (siHuR) or control-siRNA duplexes (siCtrl.) for 48 h before translation was blocked by the addition of cycloheximide (CHX, 25μg/ml). After the indicated time periods cells were harvested and extracted for total protein lysates. Graphs show means±S.D. (n=2) and depict the percentage of remaining caspase-2L protein in control-siRNA (open squares) and HuR-siRNA (filled squares) transfected cells in relation to β-actin and compared with the levels of normalized caspase-2L protein immediately before the addition of cycloheximide and set as 100%. (b, right panel) DLD-1 cells were transfected with control-siRNA (siCtrl.) or with HuR-siRNA (siHuR) and fractions of translational inactive RNPs were separated from polysomes by using sequential ultracentrifugation as described in the Materials and Methods section. The content of polysomal caspase-2 mRNA is depicted in a relationship to the amount of caspase-2 mRNA in RNP fractions from control-siRNA (open bar) and HuR-siRNA-transfected cells (black bar) and determined by semiquantitative RT-PCR. The means of two densitometrical measurements of independent experiments are shown and are depicted as relative ‘polysomes/RNPs’ ratio. (c) HuR inhibits the translation in vitro by interfering with the caspase-2-5′UTR. Plasmids containing the luciferase under the control of the 5′UTR region of caspase-2 (left panel) or the caspase-2 coding sequence without the 5′UTR (right panel) were transcribed and translated in vitro without (−), or in the presence of the indicated concentration of recombinant HuR. The translation products were subsequently detected by western blotting using the respective antibodies. The data shown represent results from two independent experiments. (d) The mTOR inhibitor rapamycin does not affect the increase in caspase-2L protein induced by HuR silencing. (left panel) DLD-1 cells were transfected as described in (B) before cells were stimulated for 24 h without (-) or with 100 nM of rapamycin (Rapa.) as indicated and harvested for total protein extraction. Thirty microgram of protein extracts were subjected to SDS-PAGE and successively probed with anti-caspase-2, anti-phospho-Ser240/244-S6 and anti-β-actin antibody. The knockdown of HuR was confirmed by probing the blot additionally with a HuR-specific antibody. (right panel) Data show densitometrical analysis of caspase-2 relative to β-actin protein in HuR-siRNA (black bars) and control-siRNA-transfected DLD-1 cells (open bars) treated without (- Rapa.) or with rapamycin (+ Rapa.). Values represent means±S.D. (n=3) P***≤0.001 versus control-siRNA transfectants that were set as 1-fold
HuR inhibits translation of caspase-2L via binding to its 5′UTR
To test whether HuR is capable of inhibiting caspase-2 at the translational level, we used a combined in vitro transcription–translation assay, in which a similar amount of rabbit reticulocyte lysate was incubated with a reporter plasmid containing the 5′UTR of caspase-2 (positioned upstream of the firefly luciferase coding sequence (CDS)) in the presence or absence of recombinant HuR. As expected, the addition of recombinant HuR had a strong inhibitory effect on the translation of luciferase independent of which amount of HuR was added (Figure 3c, left panel). To furthermore prove that the inhibitory effect on caspase-2 translation does exclusively rely on the 5′UTR, we tested whether vice versa, translation of the CDS of caspase-2 itself would be affected by HuR. However, the addition of recombinant HuR had no suppressive effect on the translation of caspase-2 if using an expression vector without the 5′UTR (Figure 3c, right panel). These data clearly indicate that the inhibitory effect of HuR on caspase-2 translation is achieved through a direct HuR binding to the 5′UTR of caspase-2.
The mTOR inhibitor rapamycin does not affect increased caspase-2 protein expression in HuR-knockdown cells
Translation of an mRNA via the 5′UTR is generally driven by either cap-dependent or by internal ribosome entry site (IRES)-mediated translation.32 We next assessed if the increase in caspase-2 protein levels observed in HuR-knockdown cells is influenced by rapamycin, an efficient inhibitor of cap-dependent translation.33 For this purpose, control-siRNA and HuR-siRNA-transfected DLD-1 cells were treated without or with rapamycin (100 nM) before time-dependent changes in the caspase-2 protein levels were monitored by western blot. The inhibitory activity of rapamycin is confirmed by the complete inhibition of the high constitutive p70S6 kinase phosphorylation indicative for a suppression of the mTOR-kinase (Figure 3d, left panel). As the increase in caspase-2 protein upon HuR silencing was not affected by rapamycin, we suggest that the modulation in caspase-2L by HuR probably relies on cap-independent translation (Figure 3d).
Transient knockdown of HuR sensitizes colon carcinoma cells to staurosporine-triggered apoptosis
To test the functional impact of the negative regulation of caspase-2 by HuR, we monitored apoptosis of DLD-1 cells by determination of sub G1 phases in HuR-depleted cells treated for 3 h without or with 250 nM staurosporine (STS), which is widely used to provoke apoptosis. In control-siRNA-transfected cells, STS caused only a weak increase in sub G1 phase entry (Figures 4a and b). By contrast, siRNA-mediated suppression of HuR sensitized cells significantly to STS-induced apoptosis (from 9.8%±1.4% to 27.9%±3.2%) (Figure 4b). Intriguingly, after the additional silencing of caspase-2 by siRNA transfection, cells showed an increased resistance toward STS-induced apoptosis (from 27.9%±3.2% back to 10.3%±0.3%), which indicates that caspase-2 is indispensable for the STS-evoked and HuR mediated increase in apoptosis (Figure 4b). In addition, silencing of caspase-2 alone had a weak inhibitory effect on the basal and STS-triggered apoptosis (Figure 4b).

Transient knockdown of HuR sensitizes colon carcinoma cells towards staurosporine-induced apoptosis. (a) Subconfluent DLD-1 cells were transfected with control-siRNA duplexes (siCtrl.) or with siRNA duplexes of HuR (siHuR), or Caspase-2 (siCasp-2), or with both in combination (siHuR+siCasp-2) for 48 h before treated for 3 h with 250 nM of staurosporine (+STS) or with vehicle before the sub G1 arrest was analyzed by Fluorescence activated cell sorting (FACS) analysis by propidium iodide (PI) staining. The intracellular PI fluorescence intensities are presented in panel (b). Values represent means±S.D. (n=3) P**≤0.01 versus the indicated siRNA transfectants from three independent experiments performed in triplicate
HuR silencing potentiates STS or α-toxin-induced caspase cleavage in adenocarcinoma cells
As a further indicator of apoptosis, we determined cleavage of caspases. As caspases exist as inactive zymogenes, which upon acitivation undergo proteolytic processing, we analyzed whether the increase in caspase-2L expression in HuR-silenced cells is accompanied by an increase in caspase-2L cleavage. However, the elevation in caspase-2 levels did not correspond with an increased caspase-2 cleavage. Importantly, the weak cleavage of caspase-2L by siRNA-mediated HuR attenuation was robustly increased if cells were treated with STS (Figure 5a). Similarly, the STS-induced processing of the effector caspase-3 was strongly enhanced after HuR silencing, whereas the total caspase-3 level was not influenced by the HuR knockdown (Figure 5a, compare lane 1 with 2). Similar changes in processing occurred with PARP, a substrate which is cleaved specifically by activated effector caspases. In contrast to caspase-2 and -3, the weak constitutive cleavage of caspase-9 was only slightly increased by STS and importantly, not enhanced by HuR silencing (Figure 5a). When additionally monitoring caspase activity by a colorimetric assay with subtype-specific substrates, we found that again, the STS-induced caspase-2 and -3 activity is most obviously seen in HuR-depleted cells (Figure 5b). In contrast, the activity of caspase-8, an initiator caspase of receptor-dependent apoptosis was not affected by HuR silencing implicating that the STS does not affect receptor-triggered apoptosis (Supplementary Figure S3).

HuR silencing potentiates staurosporine or α-toxin-induced caspase-2 and-3 cleavage in adenocarcinoma cells. (a) Cells were treated similar as described in Figure 4 before the processing of the indicated caspases and cleavage of PARP was monitored by western blot analysis. The knockdown of HuR was confirmed by probing the blot additionally with a HuR-specific antibody. Cleavage products of the indicated pro-caspases are depicted by an asterisk and molecular weights are as indicated. The blots shown represent results from three independent experiments. (b) DLD-1 cells were treated as described in panel (A) before changes in the activities of caspase-2 and caspase-3 were monitored by detection of pNA generated from the respective substrates, VDVAD-pNA or DEVD-pNA. Data represent means±S.D. (n=3) of three independent experiments performed in triplicate. P*≤0.05, P**≤0.01 compared with control-siRNA transfectants (siCtrl.) cells. (c) HeLa cells were either transfected with control (siCtrl.) or with HuR (siHuR) duplexes before cells were further challenged for 16 h without (−) or with (+) α-toxin (100 ng/ml) before harvested for total cell homogenates. Thirty microgram of protein extracts were subjected to SDS-PAGE and successively probed with antibodies raised against the indicated proteins. Cleavage products of the indicated pro-caspases are depicted by an asterisk and molecular weights are as indicated. The knockdown of HuR was confirmed by using a HuR-specific antibody and β-actin was used as a loading control. Data are representative for three independent experiments giving similar results
Next, we tested whether HuR depletion would similarly affect caspase-2-dependent apoptosis of other adenocarcinoma cell types induced by a more physiological stimulus. As a previous report could demonstrate that caspase-2 is a critical caspase in the apoptosis of HeLa cells induced by pore-forming toxins,10 we chose this model for proof-of-principle experiments. Again, caspase-2 levels reciprocally correlate with HuR protein levels (Figure 5c), demonstrating that the negative regulation of caspase-2 by HuR is not restricted to colon carcinoma cells. Importantly, when challenged with α-toxin (100 ng/ml), HuR-silenced HeLa cells exhibited a substantial increase in caspases-2 and -3 cleavage implying that HuR protects cells from α-toxin-mediated apoptosis (Figure 5c). Concomitantly, the α-toxin-induced cleavage of full-length PARP is most prominently seen in HuR-knockdown cells. In contrast, caspase-9 cleavage was not enhanced by the knockdown of HuR (Figure 5c). Together these data suggest that a reduction of HuR expression by siRNA generally sensitizes adenocarcinoma cells towards stimulus-induced apoptosis without affecting the basal rate of apoptosis.
Caspase-2L is critical for STS-triggered apoptosis
As the increase in apoptosis sensitivity by HuR knockdown could be due to the decreased expression of other HuR-regulated apoptosis regulatory proteins, the impact of caspase-2L on the increased cleavage of both proteases by HuR depletion was assessed by the additional silencing of caspase-2L. Consistent with the low stimulus-dependent accumulation in the sub G1 phase (Figure 4a), the STS-induced increase in caspase-3 and PARP cleavage was totally ablated in HuR/caspase-2L double knockdown cells and was even below the control-siRNA transfectants (Figure 6a, left panel). Consistently, silencing of caspase-2L alone showed a clear inhibitory effect on the weak caspase-3 cleavage induced by STS (Figure 6a, right panel), whereas the low constitutive caspase-9 cleavage and the STS-induced cleavage of PARP was not affected by caspase-2L silencing (Figure 6a). Unexpectedly, a quantification of western blots furthermore confirmed that the additional knockdown of caspase-2L did not only reduce, but completely reverse the HuR-dependent and STS-induced increase in the processing of both proteins (Figure 6b). Together, these data indicate that in DLD-1 cells, caspase-2L is critical for a full sensitization to stimulus-induced apoptosis by HuR depletion and mediated by activation of the executioner caspase-3 and PARP.

The critical role of caspase-2 in staurosporine-triggered apoptosis. (a) DLD-1 cells were transfected with control-siRNA duplexes (siCtrl.) or with siRNA duplexes of caspase-2L (siCasp-2) or HuR (siHuR) or alternatively, double transfected with HuR plus caspase-2L-specific siRNA duplexes (siHuR+Casp-2) for 48 h before cells were challenged with 250 nM of staurosporine (+STS) or with vehicle. Three hours after stimulation, cells were harvested for total protein extraction and the processing of the indicated caspases and PARP cleavage was detected by western blot analysis. The efficient knockdown of HuR and/or caspase-2L was confirmed by probing the blot with corresponding antibodies and β-actin was used as a loading control. The levels of cleaved caspases are depicted by an asterisk. The blots shown represent results from three independent experiments. (b) Densitometrical analysis of cleaved caspase-3 (left panel) or PARP (right panel) levels in a relationship to the contents of uncleaved caspase-3 and PARP in DLD-1 cells, which were transfected with the indicated siRNA duplexes and in response to STS (250 nM). Data represent means±S.D. (n=3) P*≤0.05, compared with control-siRNA-transfected (siCtrl.) cells and #≤0.05, ##≤0.01 compared with siHuR-transfectants (siHuR)
Discussion
In this study, we report caspase-2 as a novel target gene of the ubiquitous RNA-binding protein HuR. Emerging evidence from the last decade has implicated that HuR can efficiently promote cell survival mainly through stabilizing and/or increasing the translation of mRNAs coding for prominent antiapoptotic regulatory factors including Bcl-2,16 prothymosin (ProT) α,34 X-linked inhibitor of apoptosis35 and SIRT1.36 Conversely, under lethal stress conditions, HuR is also able to promote apoptosis mainly by increasing the expression of proapoptotic proteins as shown for p5337 and cytochrome c.38 A functional dichotomy of HuR was explained by caspase-mediated cleavage, which switches HuR from promoting cell survival to activating apoptosis.39 However, as we did not detect any cleavage products of HuR, a similar mechanism is presumably not relevant for HuR-triggered caspase-2 regulation of colon carcinoma cells (data not shown).
In contrast to the other caspases, the functional role of caspase-2 in apoptosis is less established. Notably, caspase-2 is additionally implicated in various non-apoptotic signaling pathways including cell cycle regulation, DNA-damage signaling and tumor suppression.5, 39, 40 Although, caspase-2-deficient mice develop normally and display almost no remarkable phenotypical changes, embryonic fibroblasts from these mice (MEFs) show an increased resistance toward different cytotoxic agents.9 By using RNA interference experiments, we show that HuR exerts a strong negative effect on caspase-2 expression in DLD-1 cells and a similar inverse correlation could be confirmed in SW620 (Figure 3a) and in HeLa cells (Figure 5c). The latter finding clearly indicates that the negative regulation of caspase-2L expression by HuR is not a cell type-specific phenomenon. Our findings furthermore provide further novel insights into the mechanisms underlying the broad antiapoptotic influence on epithelial cells by HuR. Given that HuR is one of few RNA-binding proteins with an exclusive stabilizing effect on target mRNA, the negative regulation of caspase-2 mRNA was somehow unexpected and is difficult to explain. Despite the fact that the 3′UTRs of the different caspase-2 splice variants bear several prototypical HuR binding sites, RNA-crosslinking and biotin pull-down assay revealed that HuR does exclusively bind to a 5′UTR commonly present in the caspase-2L (ICH-1L) encoding mRNA and in splicing variant 3 with a still unknown function (Figure 1b).
Consistent with these observations, actinomycin D experiments revealed that caspase-2 mRNA-stability is not affected by HuR (Figure 3). We therefore propose that the increase in steady-state mRNA levels of caspase-2 observed in HuR-knockdown cells may reflect a negative transcriptional mechanism due to the enhanced decay of a still unknown transcriptional repressor encoding mRNA stabilized by HuR.
In contrast, the constitutive binding of HuR to the 5′UTR of caspase-2L is a strong hint that HuR additionally confers a direct suppressive effect on caspase-2L translation. This hypothesis is corroborated by the observation that the ratio of caspase-2 mRNA abundance between polysomes and nonpolysomal RNP fractions from HuR-silenced DLD-1 cells was clearly shifted towards translational inactive RNPs when compared with the caspase-2 mRNA distribution in control-siRNA-transfected cells (Figure 3b) and further evidence for a translational regulation of caspase-2L by HuR via the 5′UTR comes from our finding that recombinant HuR had a strong suppressive effect on in vitro translation of a luciferase reporter fused to the caspase-2L-specific 5′UTR (Figure 3c), whereas the translation of caspase-2L remained unaffected by HuR if a caspase-2L expression plasmid without the caspase-2L-5′UTR. Reports on a direct repressive effect on translation by HuR are rare and have so far only been reported by several studies.23, 24, 25, 26 Noteworthy, in all of these cases, translational repression is structurally due to an interference of HuR with an IRES element residing in the 5′UTR of corresponding mRNAs.24, 25, 26 The finding that rapamycin is unable to modulate caspase-2L expression is a first hint that HuR may affect IRES-mediated translation of caspase-2L (Figure 3d). However, further experiments are needed to unveil the detailed mechanism underlying inhibition of caspase-2L translation by HuR.
Functionally, the inhibition of caspase-2L by increased HuR levels could be relevant for apoptosis resistance of tumor cells. Importantly, an increase in caspase-2L levels alone was unable to trigger apoptosis in DLD-1 cells but requires an apoptotic external stimulus (Figure 5). The fact that the HuR depletion-dependent increase in caspase-3 and PARP cleavage by STS (Figure 6) and the increased cell accumulation in the sub G1 phase (Figure 4) was almost totally impaired if caspase-2L expression was silenced by siRNA clearly demonstrates that caspase-2L is functionally relevant for STS-induced apoptosis. Similar to the effects observed with STS, the doxorubicin-induced caspase-3 cleavage was strongly enhanced after silencing of HuR (Supplementary Figure 4). In a contrast to our expectations, immunohistochemical comparison of the levels of HuR and active caspase-2 in tissue specimen from human adenocarincomas with healthy tissue did not show a clear inverse correlation of both antigens. Instead of this, a strong and intense staining of caspase-2 was observed in healthy (normal), tumor-adjacent but also in tumor tissue (Supplementary Figure S5). However, a sound evaluation of the clinical relevance of these data warrants for a statistical analysis from a larger patient cohort. Our observations furthermore suggest that caspase-2 mainly acts as an initiator rather than an executioner caspase and are in accordance to those reports that have demonstrated that in stress-induced and caspase-3-triggered apoptosis, caspase-2 can act upstream of mitochondria.41 Furthermore, previous studies could demonstrate that caspase-2 is indispensable for α-toxin-mediated apoptosis in epithelial cells and tumor suppression by mechanisms that are independent of PIDDosome formation.10, 11, 42 In summary, our data suggest that caspase-2 acts as an initiator caspase in the apoptotic signaling pathways induced by cytotoxic agents or bacterial toxins. Therefore, the constitutive inhibition of caspase-2 by HuR may constitute an additional important mechanism of the broad antiapoptotic program of adenocarcinoma cells.
Materials and Methods
Reagents
Actinomycin D (from Streptomyces species), Rapamycin and STS were obtained from Calbiochem (Schwalbach, Germany). α-toxin from S. aureus, cycloheximide, doxorubicin and the antibody against GAPDH, β-actin and β-tubulin were purchased from Sigma-Aldrich (Deisenhofen, Germany). The antibodies against caspase-2 (ICH-1, Nedd-2), and Firefly luciferase were from Millipore (Darmstadt, Germany). Rabbit antibodies against caspase-9 and against cyclin A were from Cell Signaling (Frankfurt, Germany). Antibodies raised against HuR, anti-rabbit, anti-mouse and anti-goat horseradish peroxidase linked IgG were purchased from Santa Cruz Biotechnology (Heidelberg, Germany). Antibodies raised against phospho-S6 (Ser240/244), caspase-3 and PARP were from Cell Signaling. Protein G sepharose, the ECL system and Hyperfilm were also from GE Healthcare. Ribonucleotides and modifying enzymes were from Life Technologies (Karlsruhe, Germany). All cell culture media and supplements were purchased from Life Technologies.
Cell lines and cell culture
The human colon carcinoma cell lines DLD-1 and SW620 and HeLa cells were obtained from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Cells were either grown in Dulbecco’s modified Eagle medium (DLD-1, SW620) or in RPMI medium (HeLa) supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin.
RNA interference
Epithelial cells were plated into 6-cm dishes to obtain 30–40% confluency on the next day.
siRNA transfection of subconfluent cells was performed by using the Oligofectamine reagent (Invitrogen, Karlsruhe, Germany) according to the manufacturer’s instructions.
Gene silencing was performed by transfecting 50 nM of a mixture of small interfering RNA (siRNA)-duplexes from Santa Cruz (siRNA-HuR, sc-35619) and FlexiTube siRNAs for human HuR (SI00300139, SI03166436, SI103246551 and SI03246887) or the same amount of siRNAs for caspase-2 (SI00299551) from Qiagen (Hilden, Germany). For double siRNA transfections, each siRNA was applied at 25 nM. Forty-eight hours after transfection, the cells were lysed for specific applications. The specific silencing of targeted genes was validated by western blot analysis.
Western blot analysis
Whole-cell lysates were prepared as described previously.43 Total cell lysates containing 30 μg of protein were prepared in SDS sample buffer and resolved by 12% SDS-PAGE, and transferred for immunodetection with specific primary antibodies followed by incubations with the appropriate secondary antibodies and finally visualized with chemiluminescence using an ECL system from GE Healthcare. To confirm equal loading of protein amounts, blots were re-probed with a β-actin antibody.
qRT-PCR/RT-PCR
Total RNA was extracted from whole cells by using the Tri reagent (Sigma-Aldrich). First strand cDNA was synthesized using random hexamer primer (Life Technologies) and Superscript reverse transcriptase (Life Technologies). Two-step real-time PCR was performed using a Taqman (ABI 7500) from Perkin Elmer (Rodgau, Germany). The mRNA levels for HuR, caspase-2 and β-actin were determined by using a protocol according to the ‘hot start’ real-time PCR procedure with ‘Dynamo Flash’ SYBR green (Biozym, Hessisch Oldendorf, Germany). C(T) values were normalized to the C(T) values of β-actin mRNA within the same sample.
For standard RT-PCR, the same conditions were used as described for qRT-PCR. The primer sets that were used are provided under ‘Supplementary Information’.
The PCR products (caspase-2, 279 bp; 5′UTR-Caspase-2L, 241bp; Caspase-2-v1,-v2,-v3, 2610 bp; 3′UTR-Caspase-2-v2, 394 bp; COX-2, 304 bp; cyclin A, 443 bp,) were separated on a 1% agarose gel containing 0.5 μg/ml ethidium bromide.
Immunoprecipitation
Immunoprecipitation (IP) was performed as described previously.31
RNP-IP RT-PCR analysis
Endogenous HuR-mRNA complexes were precipitated by using RNP-IP-qRT-PCR analysis as described previously.44 HuR-bound RNA was reverse transcribed (RT) using SuperScript reverse transcriptase (Life Technologies) and the levels of specific mRNAs were quantified by PCR (qRT-PCR). Normalization of input RNA was confirmed by RT reaction of total cellular RNA isolated from 10% of cell extract as was used for IP and subsequent assessment of GAPDH levels.
RNP-IP-Chip-analysis
Whole-cell lysates from siRNA experiments performed with cells from three different passages were pooled and used for RNP-IP as described previously.43 The microarray analysis was performed at the Institute for Cell Biology at the University of Essen. For microarray analysis, the quality of RNA from IP material was initially analyzed on a Agilent Bioanalyzer 2110 (Agilent Technologies, Böblingen, Germany) before mRNAs were reverse transcribed by random priming using oligo dT primers. Biotin-labeled cRNA that was generated by in vitro transcription using T7 RNA polymerase was subsequently hybridized to a Human Genome U133 Plus 2.0 GeneChip from Affymetrix (Cleveland, OH, USA) containing 47 000 transcript probes per sample. The raw microarray data were filtered by the detection of P-value ≤0.002, normalized by Z-score transformation. Changes in gene expression between different RNA groups were calculated by subtracting the average of replicate Z scores and referred to as ‘Z ratio’ (Z average in HuR IP minus Z average in IgG IP). Transcripts were considered ‘changed’ when Z ratios were ≥+1.5 or ≤−1.5. Owing to the lack of sufficient replicates, we applied simple cross-comparison studies, in which the experimental samples were pairwise compared with the respective baseline samples by using the Affymetrix MAS5 comparison algorithm. The completed list of changes in genes from array analysis is shown in the Supplementary Table. The Ingenuity Systems Pathways Analysis from Ingenuity Systems (Redwood City, CA, USA) was used to assign differentially bound transcripts to distinct network functions.
Generation of a plasmid bearing the 5′-UTR of caspase-2 for in vitro transcription
Experimental details for construction of this plasmid are provided in the ‘Supplementary information’ section.
In vitro translation assay
In vitro translation assays were performed using the TNT Coupled Reticulocyte Lysate (Promega, Mannheim, Germany), which couples in vitro transcription reaction with in vitro translation system in one single tube by following the customer’s instructions. Briefly, 200 ng of the plasmid pKS-caspase-2-5′UTR-Luc or pcDNA3-caspase-2 (cds), both of which plasmids are under the control of a T7 RNA polymerase promoter, was added directly to T7 polymerase, RNase inhibitor and amino acid mixture containing TNT Lysate reagent mix and incubated in 20 μl final reaction volume for 1.5 h at 30 °C. One or three microgram of recombinant HuR was directly added to the TNT lysate before the reaction was started. Afterwards, the translation products were directly separated by SDS-PAGE and the proteins were detected by western blot analysis using a firefly-luciferase-specific or a caspase-2-specific antibody, respectively.
Biotin pull-down assay
To generate biotinylated RNA sense probes for the biotin pull-down assay, 10 μg of either linearized plasmid pCR2.1-5′-UTR-caspase-2 or partial human reverse GAPDH (hrg) were in vitro transcribed and biotinylated by using the ‘Riboprobe Combination System Kit’ from Promega and T7 RNA polymerase and biotin-CTP (Invitrogen). Fifteen microgram of the biotinylated RNAs were conjugated to streptavidin-conjugated agarose beads in incubation buffer (10 mM Tris-HCl, pH 7.5; 150 mM KCl; 1.5 mM MgCl2; 0.5 mM DTT; 40 U/ml RNasin) at 4 °C for 2 h with continuous rotation. Subsequently, 300 μg of total cell lysates were added to the beads and incubated for 45 min at 4 °C. After intensive washing with incubation buffer, the RNA-bound proteins were eluted by addition of 300 μl of elution buffer (3 M KCl; 0.75 M MgCl2; 5 mM Tris-HCl, pH 7.5) and the pull-down material was subsequently analyzed by western blot analysis by probing the membranes successively with a HuR-specific and a β-actin-specific antibody, of which the latter one was used for an exclusion of unspecific protein binding to the RNA probe. After incubation of the secondary antibodies, the immunopositive signals were visualized by enhanced chemiluminescence.
Crosslinking-RNP-IP assay
HuR binding to specific regions of caspase-2 mRNA was analyzed by crosslinking-RNP-IP assay by using a protocol from Niranjanakumari et al.,45 with slight modifications as described previously.46
Briefly, cells were harvested by centrifugation at 100 × g for 5 min at 4 °C and resuspended in cold 0.5% formaldehyde in PBS (v/v) before being incubated at room temperature for 10 min. Crosslinking was quenched by addition of glycine (pH 7.0) to a final concentration of 0.25 M and incubated for further 5 min. After centrifugation, cells were washed and resuspended in 1 ml of RIPA buffer (50 mM Tris/HCl, pH 7.5; 1% NP-40; 0.05% SDS; 1 mM EDTA and 150 mM NaCl) supplemented with a protease inhibitor cocktail. Cell lysis was accomplished by three rounds of sonication for 20 s each. The supernatant was diluted with RIPA buffer containing RNasin and protease inhibitors, incubated with a monoclonal anti-HuR antibody (0.2 mg/ml) or, alternatively, with the same amount of mouse IgG overnight before protein G sepharose beads were added and incubated for 2 h on a rotator at 4 °C. After several washing steps with different buffers,45 the beads were collected and resuspended in 0.1 ml of 50 mM Tris-HCl, pH 7.0; 0.5 mM EDTA; 10 mM DTT and 1% SDS before incubated at 70 °C for 45 min to reverse crosslinking. Subsequently, the RNA was extracted by using the Tri reagent (Sigma-Aldrich) followed by DNase treatment. Recovered nucleic acids were precipitated with ethanol and resuspended in DEPC-treated water. The RNA was used as a template and reverse transcribed by using random hexamers and Superscript transcriptase (Life Technologies). The resulting cDNA was analyzed with the primer sets specific for different regions of the caspase-2 mRNA listed above.
Separation of polysomes from translational inactive RNPs
Trypsinized cells were collected by centrifugation for 5 min at 5000 × g at 4 °C before cell pellets pooled from eight 6-cm-culture dishes were mixed with two volumes of ice-cold lysis buffer (20 mM HEPES; 250 mM sucrose; 250 mM KCl; 5 mM MgCl2; 2 mM DTT; 1 mg/ml heparine, pH 7.5) and subsequently homogenized by freezing and thawing in liquid nitrogen followed by centrifugation at 4000 × g for 10 min at 4 °C. Supernatants were supplemented with Triton X-100 (final 1%) and 40 U/ml RNaseOUT (Invitrogen) and equal volumes (0.5 ml) loaded onto a sucrose cushion (1 M) and polysomes isolated by centrifugation at 100 000 × g for 2 h at 4 °C. Polysomal pellets were dissolved in 150 μl polysomal lysis buffer buffer. The RNA from both fractions was subsequently extracted by adding 1 ml of TRI reagent (Sigma-Aldrich) before nucleic acids were precipitated with isopropanol and resuspended in DEPC-treated water. Measurement of caspase-2L mRNA levels by RT-PCR or qRT-PCR was done as described above.
Cell cycle and apoptosis analysis
The quantification of the sub G1 population of DLD-1 cells was monitored to measure apoptosis according to a procedure published by Gong et al.47 Briefly, cells were seeded in six-well plates and transfected with the relevant siRNAs as described before. At 48 h after siRNA transfection, the cells were trypsinized, washed in PBS and stored overnight in absolute ethanol at −20 °C. After centrifugation (300 × g for 2 min), cells pellets were resuspended in 0.3 ml hypotonic buffer containing 50 μg/ml propidium iodide (Sigma-Aldrich); 0.1% sodium citrate; 0.1% Triton X-100 and 10 μg/ml RNase A for 30 min at 37 °C before measurement. Finally, cells were gated to exclude cell debris and analyzed by flow cytometry in linear mode by using the FACSDiva Software (BD, Heidelberg, Germany).
Measurement of caspase activities
The activity of caspase-2, -3 and -8 was measured by using corresponding assay kits from GenScript (Piscataway, NJ, USA) following the manufacturer’s instructions. The assay is based on spectrophotometric detection of the chromophore pnitroanilide (pNA) after cleavage from the relevant labeled caspase-specific substrate VDVAD-pNA (caspase-2), DEVD-pNA (caspase-3), and IETD-pNA (caspase-8), respectively. The pNA light emission was quantified using a microtiter plate reader at 400 nm. The relative increase of caspase activity was determined by comparing the absorbance of pNA from treated to untreated control cells.
Immunohistochemical analysis of colorectal tissue specimens
For immunohistochemical analysis the colon cancer tissue array CO 1002 (US Biomax, Rockville, MD, USA) containing paraffin-embedded tissue cores from colorectal adenocarcinomas and normal colon tissue was used and staining was performed as described previously.29 Following the endogenous peroxidase blocking step, colon cancer tissue arrays were incubated with either a rabbit polyclonal anti-HuR or with a rabbit polyclonal antibody raised against active caspase-2 from Abcam (Cambrige, UK) (both at 1 : 50). Immunohistochemistry was visualized using the DAB peroxidase substrate kit (Vector Laboratories, Lörrach, Germany). Tissue sections were counterstained with hematoxylin (Sigma-Aldrich).
Statistical analysis
Results are expressed as means±S.D. The data are presented as x-fold induction compared with untreated control (*) or compared with stimulated values (#). Statistical analysis was performed using Student’s t-test and analysis of variance (ANOVA). P–values ≤0.05 (*), (#), ≤0.01 (**), (##) and ≤0.005 (***) were considered significant.
Abbreviations
- ARE:
-
AU-rich element
- Cox-2:
-
cyclooxygenase-2
- HuR:
-
human antigen R
- PARP:
-
poly ADP-ribose polymerase
- RNP-IP:
-
ribonucleoprotein-immunoprecipitation
- STS:
-
staurosporine
- 5′-UTR:
-
5′-untranslated region
References
Kumar S . Caspase function in programmed cell death. Cell Death Differ 2007; 1: 32–43.
Riedl SJ, Shi Y . Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 2004; 11: 897–907.
Bouchier-Hayes L . The role of caspase-2 in stress-induced apoptosis. J Cell Mol Med 2010; 14: 1212–1224.
Thornberry NA, Lazebnik Y . Caspases: enemies within. Science 1998; 281: 1312–1316.
Kitevska T, Spencer DM, Hawkins CJ . Caspase-2: controversial killer or checkpoint controller? Apoptosis 2009; 14: 829–848.
Tinel A, Tschopp J . The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004; 304: 843–846.
Troy CM, Shelanski ML . Caspase-2 redux. Cell Death Differ 2003; 10: 101–107.
Baliga BC, Read SH, Kumar S . The biochemical mechanism of caspase-2 activation. Cell Death Differ 2004; 11: 1234–1241.
Bouchier-Hayes L, Oberst A, McStay GP, Connell S, Tait SW, Dillon CP et al. Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol Cell 2009; 35: 830–840.
Imre G, Heering J, Takeda AN, Husmann M, Thiede B, zu Heringdorf DM et al. Caspase-2 is an initiator caspase responsible for pore-forming toxin-mediated apoptosis. EMBO J 2012; 31: 2615–2628.
Imre G, Rajalingam K . Role of caspase-2 during pore-forming toxin-mediated apoptosis. Cell Cycle 2012; 11: 3709–3710.
Wang L, Miura M, Bergeron L, Zhu H, Yuan J . Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell 1994; 78: 739–750.
Logette E, Wotawa A, Solier S, Desoche L, Solary E, Corcos L . The human caspase-2 gene: alternative promoters, pre-mRNA splicing and AUG usage direct isoform-specific expression. Oncogene 2003; 22: 935–946.
Fushimi K, Ray P, Kar A, Wang L, Sutherland LC, Wu JY . Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc Natl Acad Sci USA 2008; 105: 15708–15713.
Iwanaga N, Kamachi M, Aratake K, Izumi Y, Ida H, Tanaka F et al. Regulation of alternative splicing of caspase-2 through an intracellular signaling pathway in response to pro-apoptotic stimuli. J Lab Clin Med 2005; 145: 105–110.
Abdelmohsen K, Lal A, Kim HH, Gorospe M . Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle 2007; 6: 1288–1292.
Nabors LB, Gillespie GY, Harkins L, King PH . HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3' untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res 2001; 61: 2154–2161.
Denkert C, Weichert W, Winzer KJ, Müller BM, Noske A, Niesporek S et al. Expression of the ELAV-like protein HuR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin Cancer Res 2004; 10: 5580–5586.
Heinonen M, Bono P, Narko K, Chang SH, Lundin J, Joensuu H et al. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res 2005; 65: 2157–2161.
Erkinheimo TL, Lassus H, Sivula A, Sengupta S, Furneaux H, Hla T et al. Cytoplasmic HuR expression correlates with poor outcome and with cyclooxygenase 2 expression in serous ovarian carcinoma. Cancer Res 2003; 63: 7591–7594.
Costantino CL, Witkiewicz AK, Kuwano Y, Cozzitorto JA, Kennedy EP, Dasgupta A et al. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res 2009; 69: 4567–4572.
López de Silanes I, Fan J, Yang X, Zonderman AB, Potapova O, Pizer ES et al. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene 2003; 22: 7146–7154.
Kullmann M, Göpfert U, Siewe B, Hengst L . ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5'UTR. Genes Dev 2002; 16: 3087–3099.
Meng Z, King PH, Nabors LB, Jackson NL, Chen CY, Emanuel PD et al. The ELAV RNA-stability factor HuR binds the 5'-untranslated region of the human IGF-IR transcript and differentially represses cap-dependent and IRES-mediated translation. Nucleic Acids Res 2005; 33: 2962–2979.
Leandersson K, Riesbeck K, Andersson T . Wnt-5a mRNA translation is suppressed by the Elav-like protein HuR in human breast epithelial cells. Nucleic Acids Res 2006; 34: 3988–3999.
Yeh CH, Hung LY, Hsu C, Le SY, Lee PT, Liao WL et al. RNA-binding protein HuR interacts with thrombomodulin 5' untranslated region and represses internal ribosome entry site-mediated translation under IL-1β treatment. Mol Biol Cell 2008; 19: 3812–3822.
Galbán S, Kuwano Y, Pullmann RJr, Martindale JL, Kim HH, Lal A et al. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1α. Mol Cell Biol 2008; 28: 93–107.
Srikantan S, Tominaga K, Gorospe M . Functional interplay between RNA-binding protein HuR and microRNAs. Curr Protein Pept Sci 2012; 13: 372–379.
Doller A, Winkler C, Azrilian I, Schulz S, Hartmann S, Pfeilschifter J et al. High-constitutive HuR phosphorylation at Ser 318 by PKCδ propagates tumor relevant functions in colon carcinoma cells. Carcinogenesis 2011; 3: 676–685.
Rübsamen D, Blees JS, Schulz K, Döring C, Hansmann ML, Heide H et al. IRES-dependent translation of egr2 is induced under inflammatory conditions. RNA 2012; 18: 1910–1920.
Doller A, Schulz S, Pfeilschifter J, Eberhardt W . RNA-dependent association with myosin IIA promotes F-actin-guided trafficking of the ELAV-like protein HuR to polysomes. Nucleic Acids Res 2013; 41: 9152–9167.
Pickering BM, Willis AE . The implications of structured 5' untranslated regions on translation and disease. Semin Cell Dev Biol 2005; 16: 39–47.
Faivre S, Kroemer G, Raymond E . Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov 2006; 5: 671–688.
Lal A, Kawai T, Yang X, Mazan-Mamczarz K, Gorospe M . Antiapoptotic function of RNA-binding protein HuR effected through prothymosin alpha. EMBO J 2005; 24: 1852–1862.
Zhang X, Zou T, Rao JN, Liu L, Xiao L, Wang PY et al. Stabilization of XIAP mRNA through the RNA binding protein HuR regulated by cellular polyamines. Nucleic Acids Res 2009; 37: 7623–7637.
Abdelmohsen K, Pullmann R Jr, Lal A, Kim HH, Galban S, Yang X et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell 2007; 25: 543–557.
Mazan-Mamczarz K, Galbán S, López de Silanes I, Martindale JL, Atasoy U, Keene JD et al. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci USA 2003; 100: 8354–8359.
Kawai T, Lal A, Yang X, Galban S, Mazan-Mamczarz K, Gorospe M . Translational control of cytochrome c by RNA-binding proteins TIA-1 and HuR. Mol Cell Biol 2006; 26: 3295–3307.
von Roretz C, Lian XJ, Macri AM, Punjani N, Clair E, Drouin O et al. Apoptotic-induced cleavage shifts HuR from being a promoter of survival to an activator of caspase-mediated apoptosis. Cell Death Differ 2013; 20: 154–168.
Dorstyn L, Puccini J, Wilson CH, Shalini S, Nicola M, Moore S et al. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ 2012; 19: 1288–1298.
Lassus P, Opitz-Araya X, Lazebnik Y . Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 2002; 297: 1352–1354.
Manzl C, Peintner L, Krumschnabel G, Bock F, Labi V, Drach M et al. PIDDosome-independent tumor suppression by Caspase-2. Cell Death Differ 2012; 19: 1722–1732.
Xin C, Ren S, Kleuser B, Shabahang S, Eberhardt W, Radeke H et al. Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-β-induced cell responses. J Biol Chem 2004; 279: 35255–35262.
Doller A, Huwiler A, Müller R, Radeke HH, Pfeilschifter J, Eberhardt W . Protein kinase Cα-dependent phosphorylation of the mRNA-stabilizing factor HuR: implications for posttranscriptional regulation of cyclooxygenase-2. Mol Biol Cell 2007; 18: 2137–2148.
Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA . Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 2002; 26: 182–190.
Doller A, Akool el-S, Huwiler A, Müller R, Radeke HH, Pfeilschifter J et al. Posttranslational modification of the AU-rich element binding protein HuR by protein kinase Cδ elicits angiotensin II-induced stabilization and nuclear export of cyclooxygenase 2 mRNA. Mol Cell Biol 2008; 28: 2608–2625.
Gong J, Traganos F, Darzynkiewicz Z . A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Anal Biochem 1994; 218: 314–319.
Acknowledgements
We are grateful to Prof. Guy Salvesen (Burnham Institute, La Jolla, USA) for providing us the plasmid pcDNA3-caspase-2. We thank Roswitha Müller and Nicole Kämpfer-Kolb for her excellent technical assistance. This work was supported by the German Research Foundation (DFG) grants, EB 257/5-1, GRK1172, RAI 739/3-1, Excellence Cluster ‘Cardiopulmonary System (ECCPS)’ EXC 147/1 and the state of Hesse (Onkogene Signaltransduktion L-4-518/55.004). KR is a PLUS-3 fellow of the Boehringer Ingelheim Foundation. AB is a scholarship holder from the ‘Deutschen Akademischen Austauschdienst (DAAD)’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Winkler, C., Doller, A., Imre, G. et al. Attenuation of the ELAV1-like protein HuR sensitizes adenocarcinoma cells to the intrinsic apoptotic pathway by increasing the translation of caspase-2L. Cell Death Dis 5, e1321 (2014). https://doi.org/10.1038/cddis.2014.279
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.279
This article is cited by
-
The regulation of protein translation and its implications for cancer
Signal Transduction and Targeted Therapy (2021)
-
Human antigen R and drug resistance in tumors
Investigational New Drugs (2019)
