
Abstract
Necroptosis is a form of programmed cell death that critically depends on RIP3 and MLKL. However, the contribution of mitochondria to necroptosis is still poorly understood. In the present study, we discovered that mitochondrial perturbations play a critical role in Smac mimetic/Dexamethasone (Dexa)-induced necroptosis independently of death receptor ligands. We demonstrate that the Smac mimetic BV6 and Dexa cooperate to trigger necroptotic cell death in acute lymphoblastic leukemia (ALL) cells that are deficient in caspase activation due to absent caspase-8 expression or pharmacological inhibition by the caspase inhibitor zVAD.fmk, since genetic silencing or pharmacological inhibition of RIP3 or MLKL significantly rescue BV6/Dexa-induced necroptosis. In addition, RIP3 or MLKL knockout mouse embryonic fibroblasts (MEFs) are protected from BV6/Dexa/zVAD.fmk-induced cell death. In contrast, antagonistic antibodies against the death receptor ligands TNFα, TRAIL or CD95 ligand fail to rescue BV6/Dexa-triggered cell death. Kinetic studies revealed that prior to cell death BV6/Dexa treatment causes hyperpolarization of the mitochondrial membrane potential (MMP) followed by loss of MMP, reactive oxygen species (ROS) production, Bak activation and disruption of mitochondrial respiration. Importantly, knockdown of Bak significantly reduces BV6/Dexa-induced loss of MMP and delays cell death, but not ROS production, whereas ROS scavengers attenuate Bak activation, indicating that ROS production occurs upstream of BV6/Dexa-mediated Bak activation. Consistently, BV6/Dexa treatment causes oxidative thiol modifications of Bak protein. Intriguingly, knockdown or knockout of RIP3 or MLKL protect ALL cells or MEFs from BV6/Dexa-induced ROS production, Bak activation, drop of MMP and disruption of mitochondrial respiration, demonstrating that these mitochondrial events depend on RIP3 and MLKL. Thus, mitochondria might serve as an amplification step in BV6/Dexa-induced necroptosis. These findings provide new insights into the role of mitochondrial dysfunctions during necroptosis and have important implications for the development of novel treatment approaches to overcome apoptosis resistance in ALL.
Similar content being viewed by others


Main
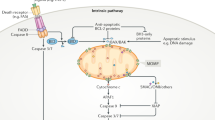
Apoptosis is one of the best characterized forms of regulated cell death which is typically characterized by the activation of caspases as cell death effector molecules.1 Besides apoptosis, necroptosis has recently been identified as another form of programmed cell death, which involves the activation of the serine/threonine kinases Receptor-Interacting Protein (RIP)1 and RIP3 and the pseudokinase mixed lineage kinase domain-like (MLKL) as key signaling molecules.2, 3, 4, 5, 6, 7 Tumor necrosis factor-α (TNFα)-mediated necroptosis represents a prototypic signaling pathway to necroptotic cell death that leads to the formation of a RIP1-RIP3-containing protein complex, referred to as the necrosome.8 Necrosome formation activates MLKL by RIP3-mediated phosphorylation, which induces MLKL oligomerization and its translocation to lipid membranes including the plasma membrane, where MLKL disrupts membrane integrity by pore formation leading to necroptotic cell death.9, 10 Caspase-8-mediated cleavage of RIP1 inhibits necroptosis,11 implying that inhibition of caspase-8 facilitates the induction of necroptosis. Although mitochondria have been implicated in necroptosis in a number of cellular systems, there is also evidence showing that mitochondria may be dispensable,12, 13 suggesting that mitochondrial contribution to necroptosis may depend on the context.
Antiapoptotic proteins like Inhibitor of Apoptosis (IAP) proteins block programmed cell death.14 X-linked IAP (XIAP) inhibits apoptosis predominately by blocking caspase activation, whereas cellular IAP (cIAP) 1 and cIAP2 proteins that harbor a really interesting new gene (RING) domain with E3 ubiquitin ligase activity promote K63-linked polyubiquitination of RIP1, leading to nuclear factor kappaB (NF-κB) activation and inhibition of cell death signaling.14 IAP proteins are expressed at aberrantly high levels in various cancers, which has been associated with poor outcome and treatment resistance.14 Treatment failure in relapsed or high-risk ALL, the most common pediatric malignancy, is associated with an unfavorable prognosis for long-term survival15, 16 and is often due to impaired cell death pathways in ALL cells, as the antileukemic activity of cytotoxic therapies critically depends on the ability of ALL cells to undergo programmed cell death.17, 18
To antagonize IAP proteins, small-molecule inhibitors such as Smac mimetics have been developed in recent years that neutralize XIAP and trigger autoubiquitination and proteasomal degradation of cIAP proteins.19 This leads to activation of non-canonical NF-κB signaling, upregulation of NF-κB target genes such TNFα and TNFα-mediated cell death in the presence of Smac mimetics.20, 21, 22 When caspase-8 activity is inhibited, Smac mimetic-stimulated depletion of cIAP protein promotes the formation of the necrosome complex.8
We previously reported that the Smac mimetic BV6 sensitizes ALL cells in vitro and in vivo for glucocorticoid-induced apoptosis.23 However, it is currently unknown whether the antileukemic activity of this Smac mimetic/glucocorticoid combination treatment is limited by defects in apoptosis pathways. In the present study, we therefore investigated the question as to whether or not BV6/Dexa cotreatment can engage non-apoptotic cell death in apoptosis-resistant ALL cells and, if so, which molecular mechanisms are involved.
Results
BV6/Dexa cotreatment induces non-apoptotic cell death in apoptosis-resistant ALL cells
We previously reported that Smac mimetics synergize with glucocorticoids to induce apoptosis in preclinical in vitro and in vivo models of ALL.23 To investigate whether this combination treatment can trigger non-apoptotic cell death in apoptosis-resistant ALL cells, we tested the effects of the Smac mimetic BV6 in combination with Dexa in the presence and absence of the broad-range caspase inhibitor zVAD.fmk. Of note, the addition of zVAD.fmk failed to protect three of the four tested ALL cell lines (i.e., Tanoue, Jurkat, KOPN-8;11) from cell death by BV6/Dexa cotreatment, whereas zVAD.fmk significantly reduced BV6/Dexa-induced cell death in Reh cells (Figure 1a). Interestingly, the analysis of key components of necroptosis and apoptosis signaling revealed RIP3 and MLKL expression in those three cell lines (i.e., Tanoue, Jurkat and KOPN-8;11) that underwent non-apoptotic cell death upon treatment with BV6/Dexa in the presence of zVAD.fmk, whereas Reh cells that were resistant to BV6/Dexa/zVAD.fmk-induced cell death lack RIP3 protein expression (Figure 1b, compare Figure 1a). Also, we discovered that Tanoue cells constitutively lack protein expression of caspase-8 (Figure 1b), and BV6/Dexa treatment did not increase caspase-8 expression in these cells (Supplementary Figure 1A).


BV6/Dexa cotreatment induces non-apoptotic cell death in apoptosis-resistant ALL cells. (a) ALL cells were treated for 24 h with BV6 and/or 200 μM Dexa in the presence or absence of 20 μM zVAD.fmk (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6; KOPN-8;11: 1 μM BV6; Reh: 300 nM BV6). Cell death was determined by FSC/SSC analysis and flow cytometry. Mean and S.D. of at least three experiments performed in triplicate are shown; *P<0.05; NS, not significant. (b) Protein levels of RIP3, MLKL and caspase-8 were assessed by western blotting, β-Actin served as loading control, asterisk indicates unspecific band. (c) Cells were treated for 18 h (Tanoue) or 12 h (Jurkat) with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and 200 μM Dexa in the presence or absence of 20 μM zVAD.fmk. Treatment of Tanoue cells with the HDAC inhibitor JNJ-26485815 served as positive control (PC). Cell death was determined by Annexin-V/PI staining and Annexin-V- and/or PI-positive or -negative cells are shown (A, Annexin-V; P, PI). Mean and S.D. of at least three experiments performed in triplicate are shown. (d) Cells were treated for 24 h with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and/or 200 μM Dexa in the presence or absence of 20 μM zVAD.fmk. Apoptosis was determined by quantification of DNA fragmentation of PI-stained nuclei using flow cytometry. Mean and S.D. of at least three experiments performed in triplicate are shown; *P<0.05
To assess apoptotic and non-apoptotic cell death, we simultaneously monitored phosphatidylserine exposure on the plasma membrane by Annexin-V in parallel with propidium iodide (PI)-staining. In Tanoue cells, BV6/Dexa predominately caused a significant increase in Annexin-V/PI double-positive cells even in the absence of zVAD.fmk (Figure 1c), indicating that this combination triggers non-apoptotic cell death in the absence of zVAD.fmk, which is consistent with lack of caspase-8 expression in Tanoue cells (Figure 1b). Treatment with the histone deacetylase (HDAC) inhibitor JNJ-26485815 was used as positive control for apoptotic cell death (Figure 1c). In Jurkat cells, the addition of zVAD.fmk to BV6/Dexa-treated cells enhanced the percentage of Annexin-V/PI double-positive cells in parallel with a reduction in Annexin-V single-positive cells (Figure 1c). This points to a switch from apoptotic to necroptotic cell death in the presence of zVAD.fmk. Control experiments confirmed very low or absent caspase-3/7 activity upon BV6/Dexa cotreatment (Supplementary Figure 1B); stimulation with TNFα and BV6 served as a positive control. Further, we analyzed DNA fragmentation as another typical feature of apoptotic cell death. BV6/Dexa cotreatment caused only a minor increase in DNA fragmentation in the absence of zVAD.fmk in Tanoue cells (Figure 1d, Supplementary Figure 1C), emphasizing that these cells undergo non-apoptotic cell death in the absence of zVAD.fmk, whereas zVAD.fmk abolished BV6/Dexa-induced DNA fragmentation in Jurkat cells (Figure 1d, Supplementary Figure 1D).
Altogether, this set of experiments demonstrates that BV6/Dexa cotreatment induces non-apoptotic cell death when caspases are inhibited (i.e., owing to the absence of caspase-8 expression or zVAD.fmk). We selected caspase-8-deficient Tanoue cells as a genetic model and zVAD.fmk-treated Jurkat cells as a pharmacological model for subsequent studies to investigate the molecular mechanisms of BV6/Dexa-induced non-apoptotic cell death.
BV6/Dexa-induced cell death upon caspase inhibition depends on RIP3 and MLKL
Caspase inhibition has previously been reported to cause a switch from apoptotic to necroptotic cell death. As the RIP1/RIP3 necrosome complex represents a central signaling platform in necroptosis, we examined whether BV6/Dexa combination treatment induces the assembly of the necrosome upon caspase inhibition. Indeed, we found an increased interaction of RIP1 and RIP3 upon BV6/Dexa cotreatment (Figure 2a), whereas BV6 or Dexa alone were unable to trigger necrosome formation (Supplementary Figure 2A). To determine whether ALL cells die via necroptosis, we inhibited key elements of the necroptotic pathway. Importantly, genetic silencing of RIP3 or MLKL by the use of two independent siRNA sequences significantly protected ALL cells from BV6/Dexa-induced cell death (Figures 2b–e). Similarly, pharmacological inhibition of RIP3 by dabrafenib or MLKL by necrosulfonamide (NSA) significantly reduced BV6/Dexa-induced cell death (Supplementary Figures 2B and C). To confirm RIP3 and MLKL as critical mediators of BV6/Dexa-induced cell death, we also used RIP3 or MLKL knockout MEFs. Consistent with our data obtained in ALL cells, BV6/Dexa-induced cell death in the presence of zVAD.fmk was significantly decreased in RIP3- or MLKL-deficient MEFs (Figures 2f–i). The protective effect in RIP3- or MLKL-deficient MEFs was comparable for BV6/Dexa/zVAD.fmk- and BV6/TNFα/zVAD.fmk-mediated cell death that was used as a well-characterized necroptotic stimulus (Supplementary Figures 2D and E). Furthermore, BV6/Dexa cotreatment caused increased phosphorylation of MLKL when caspase activation was blocked (Supplementary Figure 2F). Altogether, this set of experiments confirms that BV6/Dexa cotreatment induces necroptotic cell death when caspase activation is blocked.


BV6/Dexa-induced cell death upon caspase inhibition depends on RIP3 and MLKL. (a) Cells were treated for 6 h with BV6 (Tanoue: 3 μM; Jurkat: 5 μM), 200 μM Dexa and 20 μM zVAD.fmk. RIP3 was immunoprecipitated using anti-RIP3 antibody and detection of indicated proteins was carried out by Western blotting. β-Actin served as loading control. (b–e) ALL cells were transiently transfected with two distinct siRNAs targeting RIP3 (b and c) or MLKL (d and e) or control siRNA. Protein expression of RIP3 (b) or MLKL (d) was analyzed by western blotting, β-Actin served as loading control. Cells were treated for 24 h with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and/or 200 μM Dexa, Jurkat cells were additionally treated with 20 μM zVAD.fmk (c and e). Cell death was determined by FSC/SSC analysis and flow cytometry. Mean and S.D. of three independent experiments performed in triplicate are shown; *, P<0.05. (f and g) RIP3−/− or MLKL−/− knockout MEFs were assessed for protein levels of RIP3 (f) or MLKL (g) by western blotting in, β-Actin served as loading control. (h and i) RIP3−/− (h) or MLKL−/− (i) knockout MEFs were treated for 24 h with 5 μM BV6 and/or 200 μM Dexa in the presence of 20 μM zVAD.fmk and cell death was determined by quantification of PI-positive cells using ImageXpress Micro XLS system. Mean and S.D. of three independent experiments performed in triplicate are shown; *P<0.05
Death receptor ligands are dispensable for BV6/Dexa-induced necroptosis
As autocrine/paracrine signaling via death receptors and their ligands has been implicated in Smac mimetic-induced necroptosis,24 we investigated the requirement of death receptor ligands by using antagonistic antibodies to prevent ligand-receptor interaction. However, the TNFα-blocking antibody Enbrel failed to protect cells from BV6/Dexa-induced cell death, whereas Enbrel inhibited TNFα/BV6-induced cell death that was used as positive control (Supplementary Figures 3A and B). Similarly, tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL)- or CD95 ligand-blocking antibodies did not rescue cells from BV6/Dexa-induced necroptosis, although they blocked TRAIL- or CD95 ligand-mediated cell death (Supplementary Figures 3C–F). These results demonstrate that BV6/Dexa-induced necroptosis occurs independently of death receptor ligands.
BV6/Dexa cotreatment stimulates mitochondrial perturbations prior to cell death
Since we found death receptor ligands to be dispensable, we then asked whether mitochondrial events contribute to BV6/Dexa-induced necroptosis. To address this question, we monitored in parallel the kinetics of cell death and changes in mitochondrial functions. Interestingly, well before the onset of cell death that started around 8–12 h (Figure 3a), BV6/Dexa treatment significantly increased the percentage of cells with hyperpolarization of the MMP at very early time points up to 3 h (Figure 3b), followed by a significant loss of MMP (Figure 3c). Also, we observed a significant increase in ROS production in viable cells before they succumb to cell death using the fluorescent dye CellROX, which primarily detects superoxide radicals (Figure 3d). Similar results were obtained using MitoSOX, a fluorescent probe that selectively detects mitochondrial superoxide25 (Figure 3e). Furthermore, BV6/Dexa treatment profoundly suppressed mitochondrial respiration in viable cells that neither responded to the complex V inhibitor oligomycin thereby indicating the percentage of oxygen consumption that is utilized for ATP production nor to the uncoupler FCCP that reveals the maximal respiratory capacity of cells (Figure 3f). Altogether, this set of experiments shows that BV6/Dexa treatment perturbs mitochondrial functions during necroptosis prior to the occurrence of cell death.


BV6/Dexa cotreatment stimulates mitochondrial perturbations prior to cell death. (a) Cells were treated for indicated time points with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and/or 200 μM Dexa, Jurkat cells were additionally treated with 20 μM zVAD.fmk. Cell death was determined by FSC/SSC analysis and flow cytometry. Mean and S.D. of three independent experiments performed in triplicate are shown. (b and c) Cells were treated for indicated time points with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and/or 200 μM Dexa, Jurkat cells were additionally treated with 20 μM zVAD.fmk. MMP was assessed by TMRM staining and flow cytometry. Mean and S.D. of three independent experiments performed in triplicate are shown; *P<0.05; **P<0.01. (d and e) Cells were treated with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and/or 200 μM Dexa, Jurkat cells were additionally treated with 20 μM zVAD.fmk. ROS production was determined by CellROX staining after indicated time points (d) or MitoSOX staining after 4 h (e) and flow cytometry. Mean and S.D. of three independent experiments performed in triplicate are shown; *P<0.05. (f) Cells were treated for 6 h with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and 200 μM Dexa, Jurkat cells were additionally treated with 20 μM zVAD.fmk. Respiration was determined by the Oxygraph system (basal respiration and respiration after addition of indicated concentrations of oligomycin or FCCP). Mean and S.D. of three independent experiments are shown
BV6/Dexa-induced loss of MMP and Bak activation contributes to the kinetics of cell death
As Bax and Bak are known as critical regulators of mitochondrial events during cell death,26 we next investigated whether they are activated during BV6/Dexa-induced necroptosis. As Tanoue and Jurkat cells express no or only small amounts of Bax (Supplementary Figure 4A), we focused our studies on Bak. Interestingly, we found that Dexa and BV6 acted together to stimulate Bak activation in both ALL cell lines (Figure 4a). Monitoring of pro- and antiapoptotic Bcl-2 proteins showed that BV6/Dexa-induced Bak activation was not accompanied by altered expression levels of Bcl-2, Bcl-xL, Mcl-1 or Bim (Supplementary Figure 4B). To test the functional relevance of Bak, we silenced it using two independent siRNA sequences. Importantly, Bak knockdown significantly reduced BV6/Dexa-mediated necroptosis in a time-dependent manner, which was lost upon prolonged treatment (Figures 4b and c, Supplementary Figure 4C). Also, Bak silencing significantly decreased BV6/Dexa-stimulated loss of MMP (Figure 4d), whereas it did not protect from ROS production (Figure 4e). To investigate whether Bak dependency is specific for ALL we extended our study to additional cancer cell types. Bak knockdown significantly reduced BV6/Dexa-mediated cell death also in acute myeloid leukemia and colon carcinoma cells (Supplementary Figures 4D and E). These findings show that Bak contributes to the kinetics of BV6/Dexa-induced loss of MMP and necroptotic cell death.

BV6/Dexa-induced loss of MMP and cell death depend on Bak. (a) Cells were treated for 6 h with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and/or 200 μM Dexa, Jurkat cells were additionally treated with 20 μM zVAD.fmk. Bak activation was determined by IP using an active conformation-specific antibody. Protein expression of Bak was analyzed by western blotting. β-Actin served as loading control. (b) ALL cells were transiently transfected with two distinct siRNAs targeting Bak or control siRNA. Protein expression of Bak was analyzed by western blotting. β-Actin served as loading control. (c) Cells were treated for 24 h with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and/or 200 μM Dexa, Jurkat cells were additionally treated with 20 μM zVAD.fmk. Cell death was determined by FSC/SSC analysis and flow cytometry. Mean and S.D. of at least three independent experiments performed in triplicate are shown; *P<0.05. (d) Cells were treated for 6 h with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and 200 μM Dexa, Jurkat cells were additionally treated with 20 μM zVAD.fmk. Loss of MMP was determined by TMRM staining and flow cytometry. Mean and S.D. of three independent experiments performed in triplicate are shown; *P<0.05. (e) Cells were treated for 4 h with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and/or 200 μM Dexa, Jurkat cells were additionally treated with 20 μM zVAD.fmk. ROS production was determined by CellROX staining and flow cytometry. Mean and S.D. of three independent experiments performed in triplicate are shown; NS, not significant
ROS production is required for BV6/Dexa-induced Bak activation, loss of MMP and cell death
Our findings showing that BV6/Dexa-induced ROS production is not altered in Bak knockdown cells (Figure 4e) indicate that Bak is dispensable for ROS production. To investigate whether ROS production is required for Bak activation, we monitored Bak activation in the presence of ROS scavengers using MnTBAP, a cell-permeable superoxide dismutase (SOD) mimetic and peroxynitrite scavenger,27 or α-Tocopherol, a vitamin E derivative. Interestingly, the addition of MnTBAP or α-Tocopherol, which both significantly decreased ROS production (Supplementary Figure 5A), attenuated BV6/Dexa-stimulated Bak activation (Figure 5a). This indicates that Bak activation upon BV6/Dexa treatment occurs in a ROS-dependent manner. Therefore, we next asked whether Bak undergoes oxidative modifications in response to BV6/Dexa treatment. To address this question, we monitored oxidative thiol modifications of Bak protein using the BIAM switch assay. Intriguingly, BV6/Dexa cotreatment caused a substantial increase in oxidized Bak compared to untreated control cells (Figure 5b). Treatment with auranofin, an inhibitor of thioredoxin reductase, was used as a positive control for oxidative stress-stimulated thiol modifications (Figure 5b). Peroxiredoxin 3 (Prx3) as a marker for elevated ROS production exhibited increased levels of thiol oxidation upon both treatments (Figure 5b). In addition to attenuate Bak activation, MnTBAP or α-Tocopherol significantly reduced BV6/Dexa-induced loss of MMP and cell death (Figures 5c and d). This set of experiments indicates that ROS production is required for Bak activation, loss of MMP and cell death during BV6/Dexa-induced necroptosis.

ROS production is required for BV6/Dexa-induced Bak activation, loss of MMP and cell death. (a) Cells were treated for 6 h with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and 200 μM Dexa in the presence or absence of 100 μM MnTBAP or 100 μM α-Tocopherol, Jurkat cells were additionally treated with 20 μM zVAD.fmk. Bak activation was determined by immunoprecipitation using an active conformation-specific antibody. Protein expression of Bak was analyzed by western blotting. β-Actin served as loading control. (b) Tanoue cells were treated for 4 h with 3 μM BV6 and 200 μM Dexa, treatment with 2 μM auranofin for 40 min served as positive control for ROS-mediated oxidative modifications. Oxidative thiol modifications of Bak (oxidized) were analyzed by BIAM switch assay. Oxidized Prx3 served as marker for oxidative stress. Total protein levels of Bak and Prx3 remained unchanged upon treatment. (c) Cells were treated for 24 h with BV6 (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) and/or 200 μM Dexa in the presence or absence of 100 μM MnTBAP or 100 μM α- Tocopherol, Jurkat cells were additionally treated with 20 μM zVAD.fmk. Cell death was determined by FSC/SSC analysis and flow cytometry. Mean and S.D. of three independent experiments performed in triplicate are shown; *P<0.05. (d) Cells were treated for 8 h with BV6 and 200 μM Dexa (Tanoue: 3 μM BV6; Jurkat: 5 μM BV6) in the presence or absence of 100 μM MnTBAP or 100 μM α-Tocopherol, Jurkat cells were additionally treated with 20 μM zVAD.fmk. Loss of MMP was assessed by TMRM staining and flow cytometry. Mean and S.D. of three independent experiments performed in triplicate are shown; *P<0.05
RIP3 and MLKL are required for Bak activation and mitochondrial perturbations during BV6/Dexa-induced necroptosis
We next investigated whether mitochondrial events during BV6/Dexa-induced necroptosis are linked to the core necroptotic signaling machinery such as RIP3 and MLKL. Importantly, knockdown of RIP3 or MLKL prevented BV6/Dexa-stimulated Bak activation in apoptosis-resistant ALL cells (Figure 6a). Similarly, RIP3 or MLKL deficiency inhibited Bax activation upon BV6/Dexa treatment in MEFs (Figure 6b). Also, knockdown or deletion of RIP3 or MLKL protected ALL cells and MEFs from BV6/Dexa-triggered loss of MMP, ROS production and impairment of mitochondrial respiration (Figures 6c–h, Supplementary Figure 6). In summary, these results demonstrate that RIP3 and MLKL are required for Bak activation and mitochondrial perturbations during BV6/Dexa-induced necroptosis.


RIP3 and MLKL are required for Bak activation and mitochondrial perturbations during BV6/Dexa-induced necroptosis. Tanoue cells were transiently transfected with two distinct siRNAs targeting RIP3, MLKL or control siRNA, and treated for 6 h with 3 μM BV6 and 200 μM Dexa. MEFs were treated for 12 h with 5 μM BV6 and 200 μM Dexa in the presence of 20 μM zVAD.fmk. (a and b) Bak (a) or Bax (b) activation was determined by immunoprecipitation using active conformation-specific antibodies. Protein expression of Bak, Bax, RIP3 and MLKL were analyzed by western blotting. β-Actin served as loading control. (c and d) Loss of MMP was determined by TMRM staining and flow cytometry (c) or ImageXpress Micro XLS system (d). Mean and S.D. of three independent experiments performed in triplicate are shown; *P<0.05. (e and f) ROS production was determined by CellROX staining and flow cytometry (e) or ImageXpress Micro XLS system (f). Mean and S.D. of three independent experiments performed in triplicate are shown; *P<0.05. (g and h) Respiration was determined by the Oxygraph system. Mean and S.D. of three independent experiments performed in triplicate are shown; *P<0.05
Discussion
As necroptosis has only recently been identified as a form of programmed cell death, the underlying signaling pathways have not yet fully been understood. In the present study, we discovered that independently of death receptor ligand signaling, mitochondrial perturbations have a critical role in a model of necroptosis induced by cotreatment with the Smac mimetic BV6 and the glucocorticoid Dexa, a combination that has not yet been described before to induce necroptosis. We established that BV6/Dexa triggers necroptotic cell death, when caspase activation is blocked either due to lack of caspase-8 expression or due to pharmacological inhibition by zVAD.fmk, as genetic silencing or pharmacological inhibition of RIP3 or MLKL protect ALL cells from BV6/Dexa-induced necroptosis. The general relevance of this necroptotic model is underscored by the fact that BV6/Dexa/zVAD.fmk-triggered necroptosis is similarly impaired in RIP3 or MLKL knockout MEFs.
Mitochondria amplify BV6/Dexa-induced necroptosis
Several lines of experimental evidence emphasize the important role of mitochondrial dysfunctions during BV6/Dexa-induced necroptosis independently of signaling via death receptor ligands. First, our kinetic studies revealed that well before cells undergo cell death BV6/Dexa treatment causes mitochondrial dysfunctions including hyperpolarization of the MMP followed by loss of MMP, ROS production, Bak activation and disruption of mitochondrial respiration, indicating that these mitochondrial events contribute to the induction of necroptotic cell death.
Second, we identified Bak as a key regulator that is activated during BV6/Dexa-induced necroptosis and required for the kinetics of necroptotic cell death, as knockdown of Bak significantly delays the kinetics of BV6/Dexa-induced cell death. Bak activation likely occurs downstream of ROS production, as ROS scavengers attenuate BV6/Dexa-stimulated Bak activation, whereas Bak silencing fails to protect cells from BV6/Dexa-stimulated ROS production. In addition, we identified oxidative thiol modifications of Bak protein upon treatment with BV6/Dexa, emphasizing that ROS production contributes to Bak activation. ROS production and Bak activation likely promote the dissipation of MMP, as Bak knockdown or ROS scavengers decrease loss of MMP by BV6/Dexa.
Third, BV6/Dexa-stimulated ROS generation promotes necroptotic signaling and cell death, as neutralization of ROS by different ROS scavengers attenuates Bak activation, loss of MMP and cell death.
Fourth, all these mitochondrial alterations occur in a RIP3- and MLKL-dependent manner, since genetic knockdown of RIP3 or MLKL in ALL cells or knockout of RIP3 or MLKL in MEFs reduce BV6/Dexa-mediated ROS production, Bak activation, loss of MMP and disruption of mitochondrial respiration.
Fifth, mitochondrial perturbations occur independently of death receptor ligand signaling, since antagonistic antibodies against the death receptor ligands TNFα, TRAIL or CD95 ligand fail to rescue BV6/Dexa-triggered cell death. Altogether, these lines of evidence indicate that mitochondrial alterations are linked to the core necroptotic signaling components RIP3 and MLKL during BV6/Dexa-induced necroptosis and may serve as an amplification step.
Bak as a critical regulator of BV6/Dexa-induced necroptosis
Although pro-death proteins of the Bcl-2 family such as Bax and Bak are well known to regulate the mitochondrial pathway of apoptosis,26 their role during necroptosis is as yet poorly understood. Elimination of both Bax and Bak or, alternatively, Bak alone has been reported to protect cells from inflammation-induced necroptosis triggered by TNFα/cycloheximide/zVAD.fmk.28, 29, 30 By comparison, in the present study, we show that BV6/Dexa-induced necroptosis occurs independently of TNFα or other death receptor ligands, suggesting that the multidomain Bcl-2 family proteins Bax and Bak may have a general role in the regulation of necroptosis in response to different stimuli. During necrotic cell death, Bax and Bak as outer membrane components have been shown to promote mitochondrial swelling and rupture upon opening of the mitochondrial permeability transition pore (MPTP) by facilitating outer membrane permeability.31 Opening of the MPTP, a channel in the inner mitochondrial membrane that is composed of cyclophilin D, ANT, VDAC, results in the dissipation of the MMP, disruption of mitochondrial respiration, further ROS production, and ultimately mitochondrial swelling and rupture.32
Although in the present study we identified Bak as a key regulator of BV6/Dexa-induced necroptosis, Bax may have a role in other cell types, as the ALL cell lines that we used here harbor very low levels of Bax protein. For example, stable overexpression of an oligomerization-dead mutant form of Bax has been found to inhibit TNFα/zVAD.fmk-induced necroptosis in MEFs.30 Also, other mitochondrial proteins such as cyclophilin D may be involved, as MEFs deficient in cyclophilin D have been shown to be resistant to TNFα-induced necroptosis.2, 30
ROS contribute to necroptotic signaling and cell death
ROS generation has been implicated in contributing to necroptosis, although the source(s) and roles of ROS remain largely elusive.12, 13 Our data suggest that ROS production during BV6/Dexa-induced necroptosis may occur, at least in part, in the mitochondria, as ROS production is detected by MitoSOX, a mitochondrial superoxide selective probe, and since ROS scavengers that can neutralize mitochondrial ROS provide protection from BV6/Dexa-induced cell death. Oxidative stress has been implicated in promoting dimerization of multidomain Bcl-2 proteins such as Bax and its translocation to mitochondria.33 In line with the notion that ROS accumulation contributes to Bak activation, in the present study we identified thiol modifications of Bak protein at the onset of BV6/Dexa-mediated necroptosis and show that Bak activation occurs in a ROS-dependent manner. Furthermore, excessive ROS generation has been implied in facilitating the opening of the MPTP, which results in the dissipation of MMP and, ultimately, in the disruption of mitochondrial functions.32 Consistently, we show in the present study that ROS scavengers rescue cells from BV6/Dexa-induced drop of MMP. We previously demonstrated that ROS promote the stabilization of the RIP1/RIP3 necrosome during BV6/TNFα-induced necroptosis, thereby initiating a positive-feedback amplification loop.34 The requirement of ROS generation for necroptotic cell death has also been demonstrated in other prototypic models of necroptosis, that is, TNFα-mediated necroptosis in L929 cells or monocytes.35, 36
Although these studies underline that ROS contribute to necroptosis induced by various stimuli, other studies have argued that mitochondrial ROS are dispensable for necroptotic cell death. For example, it has been reported that Parkin-induced mitochondrial depletion prevents TNFα-induced ROS production, but not necroptotic cell death.37 Altogether, these data suggest that the requirement of ROS and mitochondria for necroptosis may depend on the cellular context.
Mitochondrial dysfunctions occurred downstream of RIP3 and MLKL
Our study indicates that mitochondrial dysfunctions may serve as an amplification step during BV6/Dexa-mediated necroptosis that is linked to the core necroptotic signaling machinery, as BV6/Dexa-stimulated ROS production, Bak activation, loss of MMP and disruption of mitochondrial respiration occurred in a RIP3- and MLKL-dependent manner. ROS production during necroptosis has previously been reported to depend on RIP33, 4 and RIP3 has been shown to interact with glutamate dehydrogenase 1, a mitochondrial matrix enzyme.4 Also, RIP1 and RIP3 were described to localize to mitochondria upon the induction of necroptosis30, 38, 39 and the translocation of necrosomes to mitochondria-associated membranes has been suggested to be essential for necroptosis signaling.40 However, the mechanistic details of how RIP kinases and MLKL may influence mitochondrial functions during necroptosis have not yet been resolved and it remains an open question whether they directly or indirectly control mitochondrial functions during necroptosis.
Taken together, our findings advance the current understanding of the role of mitochondria in necroptosis by showing that mitochondrial events serve as an amplifying step. In addition, our study has important implications for the development of novel treatment approaches to overcome apoptosis resistance in ALL by inducing necroptosis. We recently discovered that the Smac mimetic BV6 sensitizes ALL cells in vitro and in vivo for glucocorticoid-triggered apoptosis.23 Here we show that BV6/Dexa combination treatment induces necroptosis in ALL cell lines that express RIP3 protein. As the majority of cell lines derived from hematological malignancies including acute leukemia have been reported to harbor RIP3,41 acute leukemia may well be susceptible to therapeutic induction of necroptosis. Thus, Smac mimetics and glucocorticoids may offer a new approach to trigger necroptosis as an alternative form of programmed cell death in ALL.
Materials and Methods
Cell culture and chemicals
ALL cell lines were obtained from DSMZ (Braunschweig, Germany) and cultured in RPMI 1640 medium (Life Technologies, Inc., Eggenstein, Germany), immortalized MEFs were maintained in DMEM GlutaMAX-I medium (Life Technologies, Inc.). Media were supplemented with 10% fetal calf serum; Biochrom, Berlin, Germany), 1 mM pyruvate (Invitrogen, Karlsruhe, Germany), 1% penicillin/streptomycin and 25 mM HEPES (Invitrogen). The Smac mimetic BV6, which neutralizes XIAP, cIAP1 and cIAP2,20 was kindly provided by Genentech, Inc. (South San Francisco, CA, USA). The caspase inhibitor zVAD.fmk was obtained from Bachem (Heidelberg, Germany), Enbrel from Pfizer (Berlin, Germany), TNFα from Biomol (Hamburg, Germany), dabrafenib and JNJ-26485815 from Selleckchem (Houston, TX, USA), NSA from Toronto Research Chemicals Inc. (North York, CA), anti-CD95 ligand antibody from BD Pharmingen (Heidelberg, Germany), anti-TRAIL (mAb 2E5) from Enzo Life Sciences (Lörrach, Germany). CD95 ligand was kindly provided by Apoxis (Lausanne, Switzerland). Chemicals were purchased by Sigma-Aldrich (Steinheim, Germany) or Carl Roth (Karlsruhe, Germany) unless indicated otherwise.
Determination of cell death
Cell death was assessed by forward/side scatter (FSC/SSC) analysis and flow cytometry (FACS Canto II; BD Biosciences, Heidelberg, Germany) or by analyzing plasma membrane permeability with PI staining as described previously using flow cytometry42 or ImageXpress Micro XLS Widefield High-Content Analysis System (Molecular Devices, Sunnyvale, CA, USA). Apoptotic and necrotic cell death were differentiated by using Annexin-V/PI-staining (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions.
Determination of MMP, ROS production, mitochondrial respiration and activation of Bax and Bak
Bax and Bak activation was determined by immunoprecipitation (IP) as previously described.43 Briefly, cell were lysed in CHAPS lysis Buffer (10 nmol/l HEPES, pH 7.4; 150 nmol/l NaCl; 1% CHAPS). Five hundred to 1000 μg protein were immunoprecipitated and incubated overnight at 4 °C with 2 μg/ml mouse anti-Bak antibody (Ab-1; Calbiochem) or anti-Bax antibody (6A7, Sigma-Aldrich) and 10 μl pan-mouse IgG Dynabeads (Dako, Hamburg, Germany), washed with CHAPS lysis buffer and analyzed by western blotting using rabbit anti-Bak antibody or anti-Bax antibody. To determine MMP, cells were incubated with TMRM (50 nM; Invitrogen) for 30 min at 37 °C and immediately analyzed by flow cytometry or ImageXpress Micro XLS Widefield High-Content Analysis System. To analyze ROS production, cells were incubated at 37 °C for 30 min with 1 μM CellROX (Invitrogen), which primarily detects superoxide radicals according to the manufacturer's instructions, or for 10 min with 5 μM MitoSOX (Invitrogen), which detects mitochondrial superoxide,25 and immediately analyzed by flow cytometry or ImageXpress Micro XLS system. Mitochondrial respiration was measured using the Oxygraph-2k system from Oroboros (Innsbruck, Austria) as described previously.44
Western blot analysis
Western blot analysis was performed as described previously45 using the following antibodies: rabbit anti-RIP3 (Novus Biologicals, Littleton, CO, USA), rabbit anti-MLKL (GeneTex, Irvine, CA, USA, for human or Sigma-Aldrich for mouse), mouse anti-caspase-8 (Enzo Life Sciences), rabbit anti-Prx3 (Abcam, Cambridge, MA, USA), rabbit anti-Bak (BD Biosciences, rabbit anti-Bax (Millipore, Darmstadt, Germany), mouse anti-β-actin (Sigma-Aldrich) or mouse anti-GAPDH (Biotrend, Cologne, Germany) as loading control and secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Enhanced chemiluminescence was used for detection (Amersham Bioscience, Freiburg, Germany). Alternatively, secondary antibodies labeled with IRDye infrared dyes were used for fluorescence detection (Odyssey Imaging System, LI-COR Bioscience, Bad Homburg, Germany).
Gene silencing
Gene silencing by small interfering RNA (siRNA) was performed using Neon Transfection System (Invitrogen) as previously described34 using 80 nM SilencerSelect siRNAs against RIP3 (#1: s21740, #2: s21741), MLKL (#1: s47087, #2: s47088), Bak (#1 s1880, #2 s1881) or non-targeting control siRNA (no. 4390843). Cells were treated at 48 h after transfection with siRNAs.
BIAM switch assay
After treatment, cells were collected by centrifugation and resuspended in 2 ml medium. Free thiols were blocked by addition of 50 mM N-ethylmaleimide (NEM) as alkylating reagent for 5 min at room temperature in the dark. After washing proteins were precipitated in ice-cold 20% (w/v) TCA, protein pellets were washed first in 10% TCA and finally in 5% TCA.46 Proteins were resolved in 200 μl NEM-DB (8 M Urea, 5 mM EDTA, 0.5% SDS, 50 mM Tris/HCL, pH 8.5, 25 mM NEM) and incubated at 850 r.p.m. for 1 h at 37 °C in the dark. Proteins were precipitated by ice-cold acetone overnight at −20 °C, collected by centrifugation, washed in acetone, resuspended in 100 μl DTT-DB (8 M Urea, 5 mM EDTA, 0.5% SDS, 50 mM Tris/HCL, pH 8.5, 3 mM DTT) and incubated at 850 r.p.m. for 5 min at 37 °C in the dark followed by addition of 100 μl BIAM-DB (8 M Urea, 5 mM EDTA, 0.5% SDS, 50 mM Tris/HCL, pH 8.5, 10 mg/ml BIAM (EZ-Link Iodoacetyl-PEG2-Biotin, ThermoFisher Scientific)) and incubation at 850 r.p.m. for 1 h at 37 °C in the dark. Proteins were precipitated by ice-cold acetone overnight at −20 °C, collected by centrifugation, washed and resuspended in 100 μl lysis buffer (5 mM EDTA, 50 mM Tris/HCL pH 8.5, 1% Triton-X-100, 1% SDS) for 30 min on ice. Using 500 μg protein, affinity purification was performed by agarose streptavidin beads overnight at 4 °C on a wheel. After washing and collection of beads proteins were eluted by sample buffer and analyzed by western blotting.
Statistical analysis
Statistical significance was assessed by Student's t-Test (two-tailed distribution, two-sample unequal variance).
Abbreviations
- ALL:
-
acute lymphoblastic leukemia
- cIAP:
-
cellular IAP
- Dexa:
-
Dexamethasone
- FSC/SSC:
-
forward/side scatter
- HDAC:
-
histone deacetylase
- IAP:
-
Inhibitor of Apoptosis
- MEFs:
-
mouse embryonic fibroblasts
- MLKL:
-
mixed lineage kinase domain-like
- MMP:
-
mitochondrial membrane potential
- MPTP:
-
mitochondrial permeability transition pore
- NEM:
-
N-ethylmaleimide
- NF-κB:
-
Nuclear Factor kappaB
- NSA:
-
necrosulfonamide
- PI:
-
propidium iodide
- RING:
-
really interesting new gene
- RIP:
-
Receptor-Interacting Protein
- ROS:
-
reactive oxygen species
- siRNA:
-
small interfering RNA
- TNF:
-
tumor necrosis factor
- TRAIL:
-
tumor-necrosis-factor-related apoptosis-inducing ligand
- XIAP:
-
X-linked IAP
References
Taylor RC, Cullen SP, Martin SJ . Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008; 9: 231–241.
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332–336.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148: 213–227.
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA 2012; 109: 5322–5327.
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008; 135: 1311–1323.
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P . Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014; 15: 135–147.
Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014; 54: 133–146.
Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 2014; 7: 971–981.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471: 363–367.
Marshall KD, Baines CP . Necroptosis: is there a role for mitochondria? Front Physiol 2014; 5: 323.
Fulda S . Regulation of necroptosis signaling and cell death by reactive oxygen species. Biol Chem 2016; 397: 657–660.
Fulda S, Vucic D . Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov 2012; 11: 109–124.
Pui CH, Relling MV, Downing JR . Acute lymphoblastic leukemia. N Engl J Med 2004; 350: 1535–1548.
Stanulla M, Schrappe M . Treatment of childhood acute lymphoblastic leukemia. Semin Hematol 2009; 46: 52–63.
Fulda S . Tumor resistance to apoptosis. Int J Cancer 2009; 124: 511–515.
Fulda S . Therapeutic opportunities for counteracting apoptosis resistance in childhood leukaemia. Br J Haematol 2009; 145: 441–454.
Fulda S . Promises and challenges of smac mimetics as cancer therapeutics. Clin Cancer Res 2015; 21: 5030–5036.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131: 669–681.
Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007; 131: 682–693.
Wang L, Du F, Wang X . TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133: 693–703.
Belz K, Schoeneberger H, Wehner S, Weigert A, Bonig H, Klingebiel T et al. Smac mimetic and glucocorticoids synergize to induce apoptosis in childhood ALL by promoting ripoptosome assembly. Blood 2014; 124: 240–250.
Roesler S, Eckhardt I, Wolf S, Fulda S . Cooperative TRAIL production mediates IFNalpha/Smac mimetic-induced cell death in TNFalpha-resistant solid cancer cells. Oncotarget 2016; 7: 3709–3725.
Mukhopadhyay P, Rajesh M, Haskó G, Hawkins BJ, Madesh M, Pacher P . Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc 2007; 2: 2295–2301.
Fulda S, Galluzzi L, Kroemer G . Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010; 9: 447–464.
Batinic-Haberle I, Cuzzocrea S, Reboucas JS, Ferrer-Sueta G, Mazzon E, Di Paola R et al. Pure MnTBAP selectively scavenges peroxynitrite over superoxide: comparison of pure and commercial MnTBAP samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radic Biol Med 2009; 46: 192–201.
Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol Cell Biol 2011; 31: 3745–3758.
Tischner D, Manzl C, Soratroi C, Villunger A, Krumschnabel G . Necrosis-like death can engage multiple pro-apoptotic Bcl-2 protein family members. Apoptosis 2012; 17: 1197–1209.
Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM, Molkentin JD . Necroptosis interfaces with MOMP and the MPTP in mediating cell death. PLoS One 2015; 10: e0130520.
Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2013; 2: e00772.
Baines CP . The mitochondrial permeability transition pore and the cardiac necrotic program. Pediatr Cardiol 2011; 32: 258–262.
D'Alessio M, De Nicola M, Coppola S, Gualandi G, Pugliese L, Cerella C et al. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. FASEB J 2005; 19: 1504–1506.
Schenk B, Fulda S . Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene 2015; 34: 5796–5806.
Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ 2011; 18: 656–665.
Ardestani S, Deskins DL, Young PP . Membrane TNF-alpha-activated programmed necrosis is mediated by Ceramide-induced reactive oxygen species. J Mol Signal 2013; 8: 12.
Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep 2013; 5: 878–885.
Temkin V, Huang Q, Liu H, Osada H, Pope RM . Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol Cell Biol 2006; 26: 2215–2225.
Kasof GM, Prosser JC, Liu D, Lorenzi MV, Gomes BC . The RIP-like kinase, RIP3, induces apoptosis and NF-kappaB nuclear translocation and localizes to mitochondria. FEBS Lett 2000; 473: 285–291.
Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem 2013; 288: 16247–16261.
Morgan MJ, Kim YS . The serine threonine kinase RIP3: lost and found. BMB Rep 2015; 48: 303–312.
Fakler M, Loeder S, Vogler M, Schneider K, Jeremias I, Debatin KM et al. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood 2009; 113: 1710–1722.
Hacker S, Dittrich A, Mohr A, Schweitzer T, Rutkowski S, Krauss J et al. Histone deacetylase inhibitors cooperate with IFN-gamma to restore caspase-8 expression and overcome TRAIL resistance in cancers with silencing of caspase-8. Oncogene 2009; 28: 3097–3110.
Birkenmeier K, Drose S, Wittig I, Winkelmann R, Kafer V, Doring C et al. Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma are highly dependent on oxidative phosphorylation. Int J Cancer 2016; 138: 2231–2246.
Fulda S, Strauss G, Meyer E, Debatin KM . Functional CD95 ligand and CD95 death-inducing signaling complex in activation-induced cell death and doxorubicin-induced apoptosis in leukemic T cells. Blood 2000; 95: 301–308.
Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK et al. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo . Proc Natl Acad Sci USA 2008; 105: 8197–8202.
Acknowledgements
We thank D. Vucic for kindly providing BV6 and C. Hugenberg for expert secretarial assistance. This work has been partially supported by grants from Edith von Heyden-Vermächtnis (to KR), Nachlässe Maria Christine Held und Erika Hecker (to KR), Nachlass Martha Schmelz (to KR), DFG SFB815 (to SF, IW, JH, OL, KS), BMBF (to SF), Wilhelm-Sander-Stiftung (to SF) and IUAP VII (to SF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by JP Medema
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Rohde, K., Kleinesudeik, L., Roesler, S. et al. A Bak-dependent mitochondrial amplification step contributes to Smac mimetic/glucocorticoid-induced necroptosis. Cell Death Differ 24, 83–97 (2017). https://doi.org/10.1038/cdd.2016.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.102
This article is cited by
-
Immunogenic cell death in cancer: targeting necroptosis to induce antitumour immunity
Nature Reviews Cancer (2024)
-
Regulated necrosis, a proinflammatory cell death, potentially counteracts pathogenic infections
Cell Death & Disease (2022)
-
The molecular mechanism of acute liver injury and inflammatory response induced by Concanavalin A
Molecular Biomedicine (2021)
-
Necroinflammation emerges as a key regulator of hematopoiesis in health and disease
Cell Death & Differentiation (2019)
-
Ablation of RIP3 protects from dopaminergic neurodegeneration in experimental Parkinson’s disease
Cell Death & Disease (2019)
