
Abstract
Nuclear factor kappa B (NF-κB) promotes cell survival in response to genotoxic stress by inducing the expression of anti-apoptotic proteins including Bcl-xL, which protects mitochondria from stress-induced mitochondrial outer membrane permeabilization (MOMP). Here we show that the multifunctional sorting protein Pacs-2 (phosphofurin acidic cluster sorting protein-2) is required for Bcl-xL induction following DNA damage in primary mouse thymocytes. Consequently, in response to DNA damage, Pacs-2−/− thymocytes exhibit a blunted induction of Bcl-xL, increased MOMP and accelerated apoptosis. Biochemical studies show that cytoplasmic PACS-2 promotes this DNA damage-induced anti-apoptotic pathway by interacting with ataxia telangiectasia mutated (ATM) to drive NF-κB activation and induction of Bcl-xL. However, Pacs-2 was not required for tumor necrosis factor-α-induced NF-κB activation, suggesting a role for PACS-2 selectively in NF-κB activation in response to DNA damage. These findings identify PACS-2 as an in vivo mediator of the ATM and NF-κB-dependent induction of Bcl-xL that promotes cell survival in response to DNA damage.
Similar content being viewed by others


Main
The nuclear factor kappa B (NF-κB) family of dimeric transcription factors responds to effectors ranging from lipopolysaccharide (LPS) and pro-inflammatory cytokines to genotoxic agents by controlling the expression of more than 500 genes involved in immunity, inflammation, cell proliferation and cell survival.1 NF-κB is critically involved in the inhibition of apoptosis by transcribing genes encoding caspase inhibitors as well as multi-domain anti-apoptotic Bcl-2 family members such as Bcl-xL.2 Bcl-xL antagonizes multiple steps of the apoptotic program at the endoplasmic reticulum (ER)–mitochondria interface.3, 4 Notably, Bcl-xL binds proapoptotic Bax and Bak, which prevents these proapoptotic Bcl-2 members from forming pores that induce mitochondria outer membrane permeabilization (MOMP).5, 6, 7 In response to DNA damage, the tumor suppressor p53 promotes apoptosis by inducing expression of Puma, which sequesters Bcl-xL, thereby promoting Bax/Bak-mediated MOMP.8, 9
The prototypical NF-κB transcription factor, p65/p50, is tightly regulated by the inhibitory protein IκBα, which sequesters this heterodimer in the cytoplasm by masking the p65 nuclear localization signal (NLS).1, 10 Release of NF-κB from IκBα requires activation of the IκK complex. The activated (phosphorylated) IκK subunits, IκKβ and IκKα, in turn phosphorylate IκBα, marking pSer32,36-IκBα for proteasomal degradation.1, 10 In addition, activated IκKβ phosphorylates p65, promoting translocation of the transcription factor to the nucleus where it drives gene expression, including the induction of Bcl-xL.11, 12 Consistent with this model, pharmacologic inhibition of NF-κB signaling in primary thymocytes and cancer cell lines blocks Bcl-xL induction and increases cell death in response to treatment with apoptosis-inducing antibodies or exposure to DNA damage.13, 14
The canonical pathway of NF-κB activation starts with the binding of ligands, such as tumor necrosis factor-α (TNFα) or LPS, to their cognate cell surface receptors. This binding sets forth a series of intracellular signaling events leading to IκK complex activation and subsequent NF-κB activation.15 Unlike cell surface receptor-mediated NF-κB activation, much less is known about the mechanism by which DNA damage activates anti-apoptotic NF-κB signaling. Remarkably, this ‘nucleus-to-cytoplasm’ signaling critically depends on the activation of the nuclear DNA damage response kinase ataxia telangiectasia mutated (ATM), which results in the cytoplasmic activation of the IκK complex and degradation of cytoplasmic IκBα inhibitory protein.16 Current models suggest that DNA damage triggers activation of ATM in the nucleus followed by the translocation of this DNA repair kinase to the cytoplasm where it supports IκK activation.16, 17, 18, 19 However, little is known about the cell machinery involved in the ‘nucleus-to-cytoplasm’ translocation and cytoplasmic trafficking of ATM that drives DNA damage-triggered NF-κB activation.
Here we identify the multifunctional sorting protein PACS-2 (phosphofurin acidic cluster sorting protein-2) as a mediator of the ‘nucleus-to-cytoplasm’ ATM signaling required for DNA damage-induced activation of NF-κB in vivo. PACS-2 was initially identified by its role in mediating secretory pathway traffic and formation of contacts between the ER and mitochondria to regulate interorganellar communication and autophagy.20, 21, 22, 23, 24, 25, 26, 27 In response to the death ligand TRAIL (TNF-related apoptosis-inducing ligand), PACS-2 becomes dephosphorylated at Ser437, switching PACS-2 to a proapoptotic effector that drives permeabilization of mitochondria and lysosomes, which promotes activation of executioner caspases.20 Recent studies show that PACS-2 responds to DNA damage by promoting cell cycle arrest and cell survival.28, 29 In response to genotoxic stress, PACS-2 translocates to the nucleus where it inhibits SIRT1 to promote p53-dependent transactivation of p21 and p21-dependent cell cycle arrest.28 Here, we show that PACS-2 additionally supports cell survival in vivo following to DNA damage by promoting the ‘nucleus-to-cytoplasm’ ATM signaling required for NF-κB-dependent induction of Bcl-xL. Together, these findings suggest that PACS-2 coordinates multiple survival pathways in response to DNA damage.
Results
Pacs-2 mediates Bcl-xL induction and cell survival following DNA damage
The p53-dependent induction of p21-mediated cell cycle arrest following ionizing irradiation (IR) of Pacs-2−/− mice is defective in the radiosensitive gastrointestinal and hematopoietic systems (Supplementary Figure S1 and Atkins et al.28). In Pacs-2−/− mice, the reduced induction of p21 in the small intestine prevents the DNA damage-induced arrest of enterocyte migration.28 However, the effect of reduced p21 induction in Pacs-2−/− thymocytes on cell fate is unknown. We therefore exposed wild-type (WT) and Pacs-2−/− thymocytes to 5 Gy IR and measured the induction of apoptosis by flow cytometry (Figure 1a and Table 1). Apoptosis was markedly accelerated in Pacs-2−/− thymocytes, which was readily apparent as early as 4 -h post IR. Similar results were obtained by measuring caspase-3 activation (Figure 1b). Consistent with these results, loss of Pacs-2 sensitized thymocytes to DNA damage-induced MOMP, as irradiation of Pacs-2−/− thymocytes markedly increased release of mitochondrial cytochrome c into the cytosol (Figure 1c). These results suggest that Pacs-2−/− thymocytes are sensitized to DNA damage-induced apoptosis due to increased MOMP.

IR-induced apoptosis is accelerated in Pacs-2−/− thymocytes. (a) WT and Pacs-2−/− thymocytes were exposed or not to 4.5 Gy IR (6 h) and analyzed by flow cytometry for apoptotic cell death using propidium iodide/Annexin V co-staining. The frequency of early- and late apoptotic cells is shown in the lower right and upper right gates, respectively. (b) Thymocytes from WT and Pacs-2−/− mice were untreated or exposed to 5 Gy IR, harvested at the indicated times and cleaved caspase-3 measured by western blotting. Actin was used as loading control. (c) WT and Pacs-2−/− thymocytes were untreated or exposed to 5 Gy IR. At 4-h post IR, crude mitochondria and cytosol fractions were prepared and analyzed by western blotting for cytochrome c protein staining. Tubulin was used as control of cytoplasmic fraction loading
In colorectal cancer cells, siRNA knockdown of PACS-2 blunts the p53-dependent induction of p21, but not proapoptotic PUMA, which correlates with increased caspase-3 activation, albeit by 48-h post treatment.28 By contrast, exposure of Pacs-2−/− thymocytes to IR or Pacs-2−/− mice to whole-body irradiation (WBI) repressed the induction of Puma and p21 to a similar extent as determined by qPCR or western blot (Figure 2 and Supplementary Figure S1). Thus, the accelerated apoptosis in Pacs-2−/− thymocytes, despite the blunted induction of Puma, was surprising as this BH3-only protein triggers IR-induced MOMP by sequestering anti-apoptotic Bcl-2 proteins.8 We therefore asked whether the loss of Pacs-2 also suppressed induction of one or more anti-apoptotic Bcl-2 proteins, which would similarly result in increased MOMP and accelerated apoptosis. Notably, we observed a blunted induction of anti-apoptotic Bcl-xL, but not other Bcl-2 proteins, in thymocytes exposed to 5 Gy IR (Figure 2), consistent with earlier reports that Bcl-xL protects thymocytes from IR-induced cell death.30

Pacs-2 depletion alters induction of pro- and anti-apoptotic targets genes. (a) WT and Pacs-2−/− thymocytes exposed or not to 5 Gy IR (4 h) were analyzed by western blotting for the induction of the indicated protein targets. PARP cleavage was used as a marker of increased apoptosis. Actin was used as loading control. (b) WT and Pacs-2−/− mice were exposed to 5 Gy WBI and isolated thymocytes were analyzed for induction of the indicated genes by qPCR (normalized to GAPDH). Error bars represent mean±S.E.M. from four or more mice per condition. Statistical significance was determined using Student’s t-test
PACS-2 regulates DNA damage-dependent NF-κB activation
The determination that IR failed to induce Bcl-xL in Pacs-2−/− thymocytes suggested that Pacs-2 promotes NF-κB activation in response to DNA damage.2, 12, 31 Indeed, pre-treatment of isolated thymocytes with the NF-κB inhibitor Bay11-7082 prevented IR-induced Bcl-xL induction, similar to studies in tumor cell lines (Supplementary Figure S2 and Xu et al.14). We therefore tested the effect of Pacs-2 loss on IR-induced NF-κB activation. WT and Pacs-2−/− thymocytes were exposed to IR and then analyzed at increasing times for activation of the NF-κB pathway. In WT thymocytes, IR induced IκKβ activation, with maximal phosphorylation by 3-h post IR (Figure 3a). By contrast, IκKβ activation was markedly attenuated in Pacs-2−/− thymocytes and correlated with reduced pSer32,36-IκBα and stabilization of the NF-κB inhibitor (Figure 3b). Expectedly, the stabilized IκBα in irradiated Pacs-2−/− thymocytes was accompanied by a corresponding reduction of pSer536-p65 (Figure 3c) and impaired translocation of p65 to the nucleus following DNA damage (Figure 3d). Similar results were obtained analyzing thymocytes from Pacs-2−/− mice exposed to WBI (Supplementary Figure S3). Together, these data suggest that Pacs-2 is required for DNA damage-induced activation of NF-κB, which induces the expression of anti-apoptotic Bcl-xL to antagonize MOMP and promote cell survival.

Pacs-2 modulates NF-κB activation in vivo following DNA damage. (a) WT and Pacs-2−/− thymocytes were exposed to 5 Gy IR and analyzed at increasing times for the phosphorylation of IκKβ Ser176/180 by western blotting. Actin was used as loading control. (b) WT and Pacs-2−/− thymocytes were exposed to 5 Gy IR and analyzed at increasing times for the phosphorylation of IκBα at Ser32/36 and degradation (IκBα lane) by western blotting. Actin was used as protein loading control. The unspliced blots are shown in Supplementary Figure S5A. (c) Thymuses from WT and Pacs-2−/− mice were exposed to 5 Gy IR and analyzed at increasing times for the phosphorylation of pSer536-p65 by western blotting. (d) WT and Pacs-2−/− thymocytes were untreated or exposed to 5 Gy IR. At 4-h post IR, nuclear and cytosolic fractions were prepared and analyzed by western blotting for p65 distribution. Topo II and tubulin were used as markers for the nuclear and cytoplasmic fractions, respectively. p65 was quantified using AlphaView (ProteinSimple)
Pacs-2 is required for NF-κB activation induced by IR but not TNFα
Despite unrelated molecular triggers, signaling pathways that activate NF-κB in response to genomic DNA damage or ligation of cell surface receptors converge on IκK and the IκKβ-dependent phosphorylation of IκBα and p65 (for review, see Perkins32). We therefore tested whether Pacs-2 was required for NF-κB activation solely in response to DNA damage or additionally for signal transduction from cell surface receptors. We exposed WT or Pacs-2−/− thymocytes to 20 ng/ml TNFα and monitored NF-κB pathway activation. Pacs-2 loss had no effect on TNFα-induced activation of IκKβ or pSer536-p65 (Figure 4a), or formation of pSer32,36-IκBα or IκBα degradation (Figure 4b). Similarly, TNFα induced Bcl-xL to a similar extent in WT and Pacs-2−/− thymocytes by qPCR analysis (Figure 4c), suggesting that Pacs-2 is required for NF-κB activation in response to DNA damage but not by cytokines (see also Figure 3).

Pacs-2 is not required for canonical NF-κB activation. (a) WT and Pacs-2−/− thymocytes were treated with 20ng/ml TNFα and analyzed at increasing times for the phosphorylation of pSer176/180-IκKβ and pSer536-p65 by western blotting. Actin was used as protein loading control. (b) WT and Pacs-2−/− thymocytes were treated with 20ng/ml TNFα and analyzed at increasing times for the phosphorylation (pSer32/36) and degradation of IκBα by western blotting. Actin was used as the loading control. (c) WT and Pacs-2−/− thymocytes were treated with TNFα (20 ng/ml) for 30 min and analyzed for Bcl-xL induction by qPCR (normalized to GAPDH). Error bars represent mean±S.E.M. from three mice per condition. Statistical significance was determined using Student’s t-test. (d) WT and Pacs-2−/− thymocytes were treated with TNFα (20 ng/ml) for 20 min. p65 was immunoprecipitated (IP) from whole-cell lysates, and Ac-Lys310-p65 and IκBα were analyzed by western blotting. Ac-Lys310-p65 was quantified using AlphaView (ProteinSimple)
In addition to regulation by IκKβ-dependent phosphorylation, TNFα-dependent transcriptional activity of NF-κB is also regulated by SIRT1-dependent deacetylation of Lys310-p65.33 We recently reported that following DNA damage, PACS-2 inhibits SIRT1 to protect p53 acetylation and p53-dependent induction of p21.28 Thus, we were surprised that TNFα-induced transcriptional activity of NF-κB was unaffected by loss of Pacs-2 in thymocytes. Interestingly, western blot analysis of treated Pacs-2−/− thymocytes revealed reduced acetylation of Lys379-p53 but not Lys310-p65 (Supplementary Figure S1 and Figure 4d, and see Atkins et al.28), suggesting that Pacs-2 may control SIRT1 in a cell type, substrate and/or promoter-specific manner.
TNFα, but not IR, triggers PACS-2 Ser437 dephosphorylation and caspase-3 activation
TNFα signaling bifurcates at an early step to trigger both NF-κB-mediated anti-apoptotic signaling as well as Bid-dependent MOMP and caspase-3-mediated cell death. Whereas PACS-2 is required for FasL and TRAIL-induced apoptosis in cancer cell lines, the lack of effect of Pacs-2 depletion on TNFα-induced NF-κB activation led us to test whether Pacs-2 was required for TNFα-induced apoptosis. In agreement with our earlier studies, loss of Pacs-2 blocked TNFα-induced Bid cleavage as well as downstream activation of caspases-9 and -3 (Figure 5a). These findings support a role for PACS-2 in TNFα-induced apoptosis, but not TNFα-induced pro-survival signaling. Correspondingly, we found that PACS-2 Ser437 phosphorylation was increased in response to DNA damage but decreased with exposure to TNFα (Figure 5b). These findings are consistent with prior studies demonstrating that death ligands trigger dephosphorylation of PACS-2 Ser437, thereby switching PACS-2 to a proapoptotic effector mediating Bid cleavage and downstream caspase activation.20

TNFα, but not DNA, damage switches PACS-2 to become an apoptotic effector. (a) WT and Pacs-2−/− MEF cells were treated with TNFα (100 ng/ml) and cycloheximide (1 μg/ml) for the indicated times and lysates were analyzed by western blotting. (b) PACS-2-overexpressing HCT116 cells were treated with etoposide (50 μM), doxorubicin (1 μM) or human TNFα (40 ng/ml) as indicated. PACS-2 was immunoprecipitated (IP) and pSer437-PACS-2 was detected by western blotting. pSer437-PACS-2 was quantified using AlphaView (ProteinSimple). The PACS-2 and actin Mr values were determined using the MW standards. The unspliced blots are shown in Supplementary Figure S5B
The cytoplasmic pool of PACS-2 regulates the NF-κB-dependent expression of Bcl-xL
DNA damage triggers nuclear PACS-2 localization to promote p53-dependent transactivation of p21 by inhibiting the deacetylation of p53 bound to the p21 promoter.28 We therefore used a luciferase reporter assay to determine whether nuclear PACS-2 was required for promoting NF-κB-dependent Bcl-xL induction in HCT116 cells using plasmids expressing WT or NLS-deficient PACS-2 (PACS-2ΔNLS, see Atkins et al.28). In agreement with our previous study, PACS-2 WT, but not PACS-2ΔNLS, increased p21 promoter transcriptional activity (Figure 6 and Atkins et al.28). By contrast, PACS-2 WT and PACS-2ΔNLS promoted Bcl-xL induction to a similar extent, suggesting that nuclear PACS-2 mediates p53-dependent induction of p21 and cell cycle arrest, whereas cytoplasmic PACS-2 promotes NF-κB-dependent induction of Bcl-xL and cell survival.

Cytoplasmic PACS-2 mediates Bcl-xL reporter expression. HCT116 cells were transfected with a luciferase expression plasmid under the control of p21 or Bcl-xL promoter together with PACS-2 or PACS-2ΔNLS and the transcription factor p53 or p65. Cells were harvested 24 h after transfection and processed for luciferase activity. Luciferase values were normalized between samples using the signal from the co-transfected Renilla plasmid. Error bars represent mean±S.E.M. from at least three independent experiments. Statistical significance was determined using Student’s t-test
PACS-2 is required for the IR-induced cytoplasmic localization of the Atm nuclear kinase
Genotoxic stress-mediated NF-κB signaling requires activation and translocation of the DNA damage repair kinase ATM to the cytoplasm where it promotes IκK-dependent NF-κB signaling.17, 18, 34 Consistent with this model, pharmacologic inhibition of Atm prevented IR-induced IκKβ activation in mouse thymocytes (Figure 7a). To test the role of Pacs-2 in Atm-mediated ‘nucleus-to-cytoplasm’ signaling, we first compared IR-induced Atm activation in WT and Pacs-2−/− thymocytes. We found that IR-induced Atm activation (pSer1987-Atm) and substrate phosphorylation (pSer18-p53) was similar in the presence or absence of Pacs-2 (Figure 7b and Supplementary Figure S1). Next, we tested whether Pacs-2 was required for cytoplasmic translocation of Atm. We found that the levels of cytoplasmic Atm were blunted in Pacs-2−/− thymocytes following IR (Figure 7c). Similar results were observed in HCT116 cells following PACS-2 knockdown (Supplementary Figure S4).

DNA damage-induced interaction between ATM and PACS-2. (a) WT and Pacs-2−/− thymocytes were left untreated or pre-treated with ATM inhibitor KU-55933 for 1 h and then exposed to 5 Gy IR. Then, thymocytes were analyzed at increasing times for pSer176/180-IκKβ by western blotting. Actin was used as the loading control. Quantification of the Pacs-2 signal (AlphaView, ProteinSimple) revealed a 50% decrease in total Pacs-2 by 6-h post IR. (b) WT and Pacs-2−/− thymocytes were exposed to 5 Gy IR and analyzed at increasing times for pSer1987-ATM by western blotting. Actin was used as the loading control. (c) WT and Pacs-2−/− thymocytes were left untreated or exposed to 5 Gy IR. At 2-h post IR, nuclear and cytosolic fractions were prepared and ATM was detected by western blotting. Topo II and tubulin were used as nuclear and cytoplasmic markers, respectively. (d) HCT116 cells co-expressing PACS-2 and ATM were treated with etoposide (50 μM) for the indicated times. ATM was immunoprecipitated (IP), and co-precipitating PACS-2 was detected by western blotting. PACS-2 was quantified using AlphaView (ProteinSimple). Actin was used as loading control. (e) HCT116 cells co-expressing PACS-2 and ATM were treated with etoposide (50 μM) or KU-55933 or both drugs. ATM was IP and PACS-2 was detected by western blotting. PACS-2 was quantified using AlphaView (ProteinSimple). Actin was used as loading control. (f) HCT116 cells expressing PACS-2 or PACS-2/ATM were treated with or without etoposide (50 μM) as indicated, nuclear and cytoplasmic fractions were prepared, ATM was IP and co-precipitating PACS-2 was detected by western blotting. PACS-2 was quantified using AlphaView (ProteinSimple). Topo II and tubulin were used as markers for the nuclear and cytoplasmic fractions, respectively
As PACS sorting proteins interact with a number of signaling molecules, we tested whether PACS-2 could interact with ATM. HCT116 cells co-expressing ATM and PACS-2 were left untreated or subjected to DNA damage. DNA damage triggered a biphasic interaction between ATM and PACS-2 that was initially detected within 15 min, but was then reduced to background levels within 30 min of post-DNA damage (Figure 7d). A longer time course revealed that the PACS-2/ATM complex was again detected by 1-h post-DNA damage and remained stable for at least 4 h. Pretreatment of the cells with KU-55933 reduced the DNA damage-induced interaction between PACS-2 and ATM, suggesting that ATM activation promotes the ATM/PACS-2 complex formation (Figure 7e). Cell fractionation studies revealed that the PACS-2/ATM interaction was readily detected in the cytoplasmic fraction in a DNA damage-dependent manner (Figure 7f). Together, these findings suggest that PACS-2 promotes genotoxic stress-mediated NF-κB activation of anti-apoptotic Bcl-xL by interacting with ATM to promote its cytoplasmic redistribution in a KU-55933-senstive manner.
Discussion
Here we report that PACS-2 mediates the ATM and NF-κB-dependent induction of anti-apoptotic Bcl-xL in response to DNA damage. Our discovery was prompted by the observation that IR-induced apoptosis is markedly accelerated in Pacs-2−/− thymocytes, which was characterized by increased MOMP and a failure of these cells to induce mitoprotective Bcl-xL (Figures 1 and 2). Our findings are consistent with reports that IR-induced NF-κB pro-survival signaling is prominent in radiosensitive tissues and is required for induction of anti-apoptotic Bcl-2 proteins.35, 36, 37 But to our knowledge, this study is the first to uncover the mechanism that promotes this IR-induced cytoprotective pathway in vivo. Together with our report that PACS-2 mediates the p53-dependent induction of p21, these findings suggest PACS-2 coordinates p53/p21-dependent cell cycle arrest with NF-κB/Bcl-xL-dependent pro-survival signaling to support DNA repair in response to genotoxic damage.28 Indeed, the observation that ‘nucleus-to-cytoplasm’ NF-κB signaling is required for the induction of p21 and prolonged cell cycle arrest supports this possibility.38
This study, together with our recent report,28 reveals that PACS-2 uses distinct mechanisms to mediate the p53/p21 and NF-κB/Bcl-xL cytoprotective pathways. In response to DNA damage, a pool of PACS-2 translocates to the nucleus where it binds and inhibits the NAD+-dependent deacetylase SIRT1 to increase p53 acetylation on the p21 promoter and, consequently, transcriptional output of p21.28 By contrast, we report here that nuclear PACS-2 is not required for NF-κB-dependent induction of Bcl-xL (Figure 6). Rather, cytoplasmic PACS-2 promotes NF-κB signaling in response to DNA damage at an early step required for redistribution of ATM to the cytoplasm where it triggers IκK activation (Figure 3). The DNA damage-dependent interaction between cytoplasmic PACS-2 and ATM supports this possibility (Figure 7). This biphasic PACS-2/ATM interaction is similar to that reported for the interaction between ATM and BRAT1/BAAT1, which, similar to PACS-2, has roles in the nucleus and cytoplasm.39, 40 Whether these biphasic interactions impact compartment- or temporal-specific roles of ATM partners in resolving DNA damage is unknown. The marked reduction of Pacs-2 levels by 6-h post IR (Figures 3a and c and 7a) suggests that the role of this multifunctional protein is temporally regulated in thymocytes. Although we do not yet know the mechanism controlling Pacs-2 levels following DNA damage, studies in hepatocarcinoma cells show that PACS-2 is targeted for proteasomal degradation by cellular inhibitor of apoptoses.41 Thus, whether the reduced levels of Pacs-2 following IR reflect elimination of a cytoprotective role for Pacs-2 or, conversely, shielding from its proapoptotic functions remain to be determined.20, 28
Our determination that loss of Pacs-2 repressed DNA damage-dependent NF-κB activation but had no apparent role in the canonical cell surface NF-κB activation pathway was initially surprising given the role of SIRT1 in the repression of NF-κB activation.33 Western blot analysis, however, showed that Pacs-2 was required for inhibiting the SIRT1-mediated deacetylation of p53, but not p65, in thymocytes (Figure 4). The lack of effect of PACS-2 on SIRT1-regulated NF-κB activation may result from a substrate or context-specific role for PACS-2 in SIRT1 regulation. Indeed PACS-2 binds SIRT1 near a region that interacts with allosteric regulators, suggesting that PACS-2 may inhibit SIRT1 in a substrate or promoter-specific manner (28, 42 and Figure 2).
ATM is a central regulator of the DNA damage response,43 which, similar to PACS-2, responds to genotoxic stress by coordinating DNA repair and cell cycle arrest pathways inside nucleus with the activation of pro-survival NF-κB pathway in the cytoplasm.17, 18, 19, 43 Following DNA damage, a small pool of nuclear ATM translocates to the cytoplasm where it initiates formation of signalosome complexes required for IκK-mediated NF-κB activation.16, 28, 31, 43 However, little is known about the mechanism of the ATM ‘nucleus-to-cytoplasm’ translocation and its cytoplasmic trafficking to drive NF-κB activation in response to DNA damage. Current models suggest that DNA damage increases nuclear export of ATM into the cytoplasm.44, 45 However, our determination that PACS-2 is required for redistribution of ATM to the cytoplasm and that cytoplasmic PACS-2 interacts with ATM suggests that PACS-2 may instead redistribute ATM to the cytoplasm by impeding retrieval to the nucleus, possibly by regulating one or more trafficking steps required for NF-κB signalosome formation and activation, and not by accelerating nuclear export (Figure 7). This model suggests that ATM undergoes basal nucleocytoplasmic trafficking and would thus have roles in multiple compartments. In support of this possibility, cytoplasmic ATM localizes to membrane compartments where it regulates endocytosis, membrane traffic and cytoskeletal dynamics.46, 47, 48, 49, 50
Similar to TNF receptors, cell surface EGFR ligation can trigger NF-κB signaling.51 In response to DNA damage, however, EGFR translocates to the nucleus where it directly activates ATM at Tyr370,52 raising the possibility that EGFR may also promote NF-κB signaling by the ATM-dependent ‘nucleus-to-cytoplasm’ pathway. Recent studies show that PACS-2 promotes EGFR signaling in vivo by regulating the endosomal trafficking of the metalloproteinase ADAM17.23 Consequently, EGFR signaling is blunted in Pacs-2−/− mice. These findings raise the possibility that despite the similar extent of ATM activation in WT and Pacs-2−/− thymocytes, as determined by pSer1987 staining, it is possible that loss of Pacs-2 may indirectly affect one or more post-translational modifications on ATM necessary for DNA repair or perhaps nucleus-to-cytoplasm trafficking.
PACS-2 is a member of an emerging collection of proteins with roles in regulating both membrane traffic and nuclear gene expression.53 The PACS genes appeared first in metazoans where they have early- and conserved roles in secretory pathway traffic.22, 23, 54, 55, 56, 57 Sequence alignment and functional analyses reveal that the vertebrate PACS proteins underwent evolutionary adaptation with the acquisition of nuclear-trafficking motifs and, in the case of PACS-2, an Akt phosphorylation site at Ser437.57 The acquisition of nuclear-trafficking functions in PACS-2 coincided with the ability of vertebrate p53 to direct cytoprotective p21-dependent cell cycle arrest in addition to its evolutionarily conserved proapoptotic functions.28, 58 The phosphorylation state of PACS-2 Ser437 serves as a molecular switch between cellular homeostasis and apoptosis. Whereas Akt phosphorylated PACS-2 mediates homeostatic membrane traffic, death ligands trigger dephosphorylation at PACS-2 Ser437, which then promotes translocation of proapoptotic Bid to mitochondria and execution of apoptosis.20, 27 Thus, our finding that DNA damage stabilizes PACS-2 phosphorylation is consistent with a cytoprotective role for PACS-2 in coordinating the p53/p21 and NF-κB/Bcl-xL pathways (Figure 5). Future analyses of the PACS proteins are expected to provide further insight into the complex pathways that control cell- and tissue homeostasis.
Materials and Methods
Experimental animals
The University of Pittsburgh, School of Medicine, approved all animal studies. Isogenic WT and Pacs-2−/− C57BL/6 mice were maintained as described.20 Thymocytes were released from WT and Pacs-2−/− thymus by using the frosted ends of sterile microscope slides and suspended in complete DMEM media. Released thymocytes were counted and equal number of cells were exposed to IR using a Cesium source irradiator. Where indicated in the Figure legends, WT and Pacs-2−/− mice were exposed to 5 Gy WBI using a J.L. Shepherd & Associates Mark I Model 30 cesium irradiator (San Fernando, CA, USA) (1.475 Gy/min) in a Plexiglas pie plate rotating at 7 r.p.m. Isolated thymuses were washed twice in PBS and processed as described in the Figure legends.
Antibodies, chemicals and plasmids
The following antibodies and reagents were obtained as indicated: PACS-2 (193),20, 24 PUMA,59 Chemicon, Billerica, MA, USA; actin MAB1501), Novocastra (Buffalo Grove, IL, USA) (p53 CM5 #NCL-p53-CM5p), Cell Signaling Technology (Danvers, MA, USA) (phospho-p65Ser536 #3033, phospho-IκKα/βSer176/180 #2697, phospho-IκBαSer32/36 #9246, IκBα#9242: acetylp53Lys379 #2570, phospho-p53Ser18 #9284, cleaved caspase-3 #9664, PARP (46D11) #9532, α/β-tubulin #2148, Bcl-xL #2764, Bcl-2 #3498, Mcl-1 #5453, Bak #12105, caspase-9 #9508, acetyl-NF-κB p65 (Lys310) #3045), Santa Cruz Biotechnology (Dallas, TX, USA) (p65 #sc-372, IκKα/β #sc-7607, Bax N-20 #sc-493, HA Y-11 #sc-805), BD Pharmingen (San Jose, CA, USA) (p21 #556430, Topo II beta #611492, cytochrome c #556433), Sigma-Aldrich (St. Louis, MO, USA) (Flag antibody #F7425, FLAG mAb M2-agarose #A2220, doxorubicin #D1515, etoposide #E1383, cycloheximide #C1988, human TNFα #T6674, Neocarzinostatin (NCS) #N9162, Bay11-7082 #B5556), Covance (Emeryville, CA, USA) (HA mAb clone HA.11 MMS-101 R), R&D Systems (Minneapolis, MN, USA) (Bid/tBid #MAB860, mouse TNFα #410-MT-010), Pierce (Rockford, IL, USA) (GFP tag #MA5-15256). Protein G sepharose 4 fast flow was obtained from GE Healthcare (Marlborough, MA, USA) (#17-0618-01). Phospho-Akt substrate rabbit monoclonal antibody #81E12B5X (Cell Signaling Technology) was used to detect phosphorylation of pSer437-PACS-2. Anti-p50 antibody was obtained from Dr Gutian Xiao (UPCI, Pittsburgh, PA, USA), anti-ATM and pSer1987-Atm (mouse) were from Dr Christopher Bakkenist (UPCI, Pittsburgh, PA), and anti-pSer1981-ATM (human) and KU-55933 were from Dr Li Lan (UPCI).
Plasmids used were obtained as indicated. p53-Flag (#10838) and p65-Flag (#20012) were obtained from Addgene (Cambridge, MA, USA), and pRL-TK (#E2241) was from Promega (Madison, WI, USA). HA-tagged PACS-2 and PACS-2ΔNLS were described previously.28 pBcl-xL luciferase reporter was a gift from Dr Satya Narayan (University of Florida, Gainesville, FL, USA), p21 luciferase reporter plasmid was a gift from Dr Bert Vogelstein (Johns Hopkins Kimmel Cancer Center, Baltimore, MD, USA), and pYFP-Flag-ATM was a gift from Dr David J. Chen (University of Texas Southwestern Medical Center, Dallas, TX, USA).
Cell harvest and tissue processing
WT and Pacs-2−/− thymocytes were cultured in DMEM+10% FBS and 0.5 mM 2-mercaptoethanol, irradiated with 5 Gy and harvested in mRIPA (50 mM Tris (pH 8.0), 150 mM NaCl, 1% NP-40, 1% deoxycholate, protease inhibitors (0.5 mM PMSF, 0.1 μM aprotinin, E-64 and leupeptin) and phosphatase inhibitors (1 mM Na3VO4 and 20 mM NaF)). Mouse tissues were homogenized in RIPA (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 1% deoxycholate and 0.1% SDS) containing protease/phosphatase inhibitors using a motorized Teflon glass homogenizer. Protein concentration was determined using the Bradford assay according to manufacturer’s instructions (Bio-Rad, Hercules, CA, USA; #500-0006). Where indicated, p65 was immunoprecipitated from thymocytes lysates with anti-p65 antibody overnight at 4 °C, immune complexes were captured with protein G sepharose for 2 h at 4 °C, washed three times in lysis buffer and eluted in SDS sample buffer before western blotting. For PACS-2/ATM co-immunoprecipitation, cells were harvested in 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40 and protease/phosphatase inhibitors. ATM was immunoprecipitated with anti-Flag antibody (mAb M2) overnight at 4oC, immune complexes were captured with protein G sepharose beads, washed three times in PBS and 1% NP-40, and eluted in SDS sample buffer before western blotting. Western blots were developed with Pierce ECL Western Blotting Substrate (ThermoFisher, Waltham, MA, USA) using the Protein Simple FluorChem E image acquisition system (San Jose, CA, USA) and signals were quantified using the AlphaView image analysis software package (ProteinSimple, San Jose, CA, USA).
Flow cytometry analyses
Thymocytes were collected in DMEM supplemented with 10% FBS and 0.5 mM 2-mercaptoethanol, irradiated with 4.5 Gy (4 h), and stained using Annexin V-FITC apoptosis kit (Calbiochem, Billerica, MA, USA; #PF032) according the manufacturer’s instructions. Flow cytometry was performed on a FACSCalibur (BD, San Jose, CA, USA) by listmode acquisition using CellQuest acquisition/analysis software (BD). Data were analyzed using FCS Express (Glendale, CA, USA) (v 3.0).
Cell fractionation
For nuclear and cytoplasmic fractionation, cells were washed twice in ice-cold PBS. Then, the cells were harvested in 400 μl buffer 1 (50 mM Tris (pH 7.9), 10 mM KCl, 1 mM EDTA, 0.05% NP-40, 10% glycerol and protease/phosphatase inhibitors) and centrifuged at 6000 r.p.m. for 3 min at 4 °C. The nuclear pellet was lysed with 150μl buffer 2 (20 mM HEPES (pH 7.9), 400 mM NaCl, 10 mM KCl, 1% NP-40, 20% glycerol, 1 mM EDTA and protease/phosphatase inhibitors) for 20 min at 4 °C, then centrifuged at 14 000 r.p.m. for 10 min. Equal amount of proteins were loaded in western blot. For mitochondria–cytoplasm fractionation, untreated or IR thymocytes as indicated were washed twice in ice-cold PBS. Then, thymocytes were dounce-homogenized in mitochondrial buffer (250 mM sucrose, 10 mM Tris (pH 7.50), 1 mM EGTA and protease/phosphatase inhibitors) using a Teflon douncer and centrifuged at 3600 r.p.m. for 5 min at 4 °C to pellet nuclei and unbroken cells. Supernatant was transferred to a fresh tube and centrifuged again at 14 000 r.p.m. for 10 min at 4 °C to separate the mitochondrial (pellet) and cytoplasmic (supernatant) fractions. Equal amount of proteins were loaded in western blot.
Luciferase assays
HCT116 cells were co-transfected with pRL-TK Renilla and either the p21- or Bcl-xL luciferase reporter plasmids. At 24 h post transfection, samples were processed using the Dual-Luciferase Reporter Assay System (Promega) according to manufacturer’s instructions. Firefly and Renilla luciferase activities were measured using a Synergy 2 (BioTek, Winooski, VT, USA). A ratio between Firefly and Renilla signals was determined for each sample and normalized to the control.
qRT-PCR
WT and Pacs-2−/− mice exposed to 5 Gy WBI and RNA were isolated 6-h post IR using RNeasy according to manufacturer’s instructions (#74104; Qiagen, Valencia, CA, USA). RNA was reverse transcribed using Superscript III first strand cDNA synthesis kit (#18080-051; Invitrogen, Waltham, MA, USA). For TNFα experiments, isolated thymocytes were incubated with 20 ng/ml mouse TNFα for 30 min and then processed for qPCR. For Bay11-7280 experiments, isolated thymocytes were pre-incubated with 10 μM Bay11-7082 for 1 h, irradiated and then processed for qPCR 3-h post IR. Reactions were performed in an ABI StepOne Real-Time PCR System with the Power SYBR GREEN PCR Master Mix (#4368706; Applied Biosystems, Waltham, MA, USA) using the following primer pairs. All reactions were performed in triplicate.
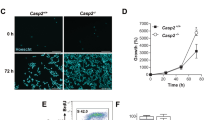
Apoptotic induction in Pacs-2−/− MEFs
SV40-transformed WT and Pacs-2−/− MEFs were seeded into 35 mm2 plates 1 day before experiment. Cells were treated with 100 ng/ml recombinant mouse TNFα (R&D Systems) and 1 μg/ml cycloheximide (CHX). Cells were harvested directly into heated 1x SDS sample buffer at 0, 6 and 12 h post treatment. Cell lysates were sonicated and western blotted for apoptotic markers.
Abbreviations
- ATM:
-
ataxia telangiectasia mutated
- ER:
-
endoplasmic reticulum
- IR:
-
ionizing radiation
- LPS:
-
lipopolysaccharide
- MOMP:
-
mitochondrial outer membrane permeabilization
- NF-κB:
-
nuclear factor kappa light-chain enhancer of activated B cells
- NLS:
-
nuclear localization signal
- PACS-2:
-
phosphofurin acidic cluster sorting protein-2
- TNFα:
-
tumor necrosis factor-α
- TRAIL:
-
tumor necrosis factor-related apoptosis-inducing ligand
- WBI:
-
whole-body irradiation
References
Napetschnig J, Wu H . Molecular basis of NF-kappaB signaling. Annu Rev Biophys 2013; 42: 443–468.
Chen C, Edelstein LC, Gelinas C . The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol Cell Biol. 2000; 20: 2687–2695.
Thomenius MJ, Distelhorst CW . Bcl-2 on the endoplasmic reticulum: protecting the mitochondria from a distance. J Cell Sci 2003; 116 (Pt 22): 4493–4499.
Eno CO, Eckenrode EF, Olberding KE, Zhao G, White C, Li C . Distinct roles of mitochondria- and ER-localized Bcl-xL in apoptosis resistance and Ca2+ homeostasis. Mol Biol Cell 2012; 23: 2605–2618.
Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW . Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008; 6: e147.
Li C, Fox CJ, Master SR, Bindokas VP, Chodosh LA, Thompson CB . Bcl-X(L) affects Ca(2+) homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proc Natl Acad Sci USA 2002; 99: 9830–9835.
Distelhorst CW, Bootman MD . Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: role in Ca(2+) signaling and disease. Cell Calcium 2011; 50: 234–241.
Yu J, Zhang L . PUMA, a potent killer with or without p53. Oncogene 2008; 27 (Suppl 1): S71–S83.
Follis AV, Chipuk JE, Fisher JC, Yun MK, Grace CR, Nourse A et al. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat Chem Biol 2013; 9: 163–168.
Xiao G, Fu J . NF-kappaB and cancer: a paradigm of Yin-Yang. Am J Cancer Res 2011; 1: 192–221.
Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W . IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 1999; 274: 30353–30356.
Israel A . The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol 2010; 2: a000158.
Khoshnan A, Tindell C, Laux I, Bae D, Bennett B, Nel AE . The NF-kappa B cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes. J Immunol 2000; 165: 1743–1754.
Xu RX, Liu RY, Wu CM, Zhao YS, Li Y, Yao YQ et al. DNA damage-induced NF-kappaB activation in human glioblastoma cells promotes miR-181b expression and cell proliferation. Cell Physiol Biochem 2015; 35: 913–925.
Vallabhapurapu S, Karin M . Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009; 27: 693–733.
Wu ZH, Shi Y, Tibbetts RS, Miyamoto S . Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 2006; 311: 1141–1146.
Hinz M, Stilmann M, Arslan SC, Khanna KK, Dittmar G, Scheidereit C . A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol Cell 2010; 40: 63–74.
Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S et al. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell 2010; 40: 75–86.
Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S et al. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol Cell Biol 2011; 31: 2774–2786.
Aslan JE, You H, Williamson DM, Endig J, Youker RT, Thomas L et al. Akt and 14-3-3 control a PACS-2 homeostatic switch that integrates membrane traffic with TRAIL-induced apoptosis. Mol Cell 2009; 34: 497–509.
Atkins KM, Thomas L, Youker RT, Harriff MJ, Pissani F, You H et al. HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation: analysis using short interfering RNA and knock-out mice. J Biol Chem 2008; 283: 11772–11784.
Dikeakos JD, Thomas L, Kwon G, Elferich J, Shinde U, Thomas G . An interdomain binding site on HIV-1 Nef interacts with PACS-1 and PACS-2 on endosomes to down-regulate MHC-I. Mol Biol Cell 2012; 23: 2184–2197.
Dombernowsky SL, Samsoe-Petersen J, Petersen CH, Instrell R, Hedegaard AM, Thomas L et al. The sorting protein PACS-2 promotes ErbB signalling by regulating recycling of the metalloproteinase ADAM17. Nat Commun 2015; 6: 7518.
Kottgen M, Benzing T, Simmen T, Tauber R, Buchholz B, Feliciangeli S et al. Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J 2005; 24: 705–716.
Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K et al. The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell 2008; 19: 2777–2788.
Werneburg NW, Bronk SF, Guicciardi ME, Thomas L, Dikeakos JD, Thomas G et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein-induced lysosomal translocation of proapoptotic effectors is mediated by phosphofurin acidic cluster sorting protein-2 (PACS-2). J Biol Chem 2012; 287: 24427–24437.
Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 2005; 24: 717–729.
Atkins KM, Thomas LL, Barroso-Gonzalez J, Thomas L, Auclair S, Yin J et al. The multifunctional sorting protein PACS-2 regulates SIRT1-mediated deacetylation of p53 to modulate p21-dependent cell-cycle arrest. Cell Rep 2014; 8: 1545–1557.
Barroso-Gonzalez J, Thomas G . Endosome traffic machinery meets the p53-p21 axis. Mol Cell Oncol 2015; 2: e975075.
Chao DT, Linette GP, Boise LH, White LS, Thompson CB, Bcl-XL Korsmeyer SJ . and Bcl-2 repress a common pathway of cell death. J Exp Med 1995; 182: 821–828.
Karin M . How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene 1999; 18: 6867–6874.
Perkins ND . Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 2007; 8: 49–62.
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004; 23: 2369–2380.
Li N, Banin S, Ouyang H, Li GC, Courtois G, Shiloh Y et al. ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. J Biol Chem 2001; 276: 8898–8903.
Zhou D, Brown SA, Yu T, Chen G, Barve S, Kang BC et al. A high dose of ionizing radiation induces tissue-specific activation of nuclear factor-kappaB in vivo. Radiat Res 1999; 151: 703–709.
Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci USA 2004; 101: 2452–2457.
Wang Y, Meng A, Lang H, Brown SA, Konopa JL, Kindy MS et al. Activation of nuclear factor kappaB In vivo selectively protects the murine small intestine against ionizing radiation-induced damage. Cancer Res 2004; 64: 6240–6246.
Wuerzberger-Davis SM, Chang PY, Berchtold C, Miyamoto S . Enhanced G2-M arrest by nuclear factor-{kappa}B-dependent p21waf1/cip1 induction. Mol Cancer Res 2005; 3: 345–353.
So EY, Ouchi T . Functional interaction of BRCA1/ATM-associated BAAT1 with the DNA-PK catalytic subunit. Exp Ther Med 2011; 2: 443–447.
So EY, Ouchi T . BRAT1 deficiency causes increased glucose metabolism and mitochondrial malfunction. BMC Cancer 2014; 14: 548.
Guicciardi ME, Werneburg NW, Bronk SF, Franke A, Yagita H, Thomas G et al. Cellular inhibitor of apoptosis (cIAP)-mediated ubiquitination of phosphofurin acidic cluster sorting protein 2 (PACS-2) negatively regulates tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) cytotoxicity. PLoS One 2014; 9: e92124.
Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013; 339: 1216–1219.
Shiloh Y, Ziv Y . The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14: 197–210.
Berchtold CM, Wu ZH, Huang TT, Miyamoto S . Calcium-dependent regulation of NEMO nuclear export in response to genotoxic stimuli. Mol Cell Biol 2007; 27: 497–509.
McCool KW, Miyamoto S . DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol Rev 2012; 246: 311–326.
Ambrose M, Goldstine JV, Gatti RA . Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum Mol Genet 2007; 16: 2154–2164.
Ismail F, Ikram M, Purdie K, Harwood C, Leigh I, Storey A . Cutaneous squamous cell carcinoma (SCC) and the DNA damage response: pATM expression patterns in pre-malignant and malignant keratinocyte skin lesions. PLoS One 2011; 6: e21271.
Lim DS, Kirsch DG, Canman CE, Ahn JH, Ziv Y, Newman LS et al. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc Natl Acad Sci USA 1998; 95: 10146–10151.
Watters D, Kedar P, Spring K, Bjorkman J, Chen P, Gatei M et al. Localization of a portion of extranuclear ATM to peroxisomes. J Biol Chem 1999; 274: 34277–34282.
Zhang L, Tie Y, Tian C, Xing G, Song Y, Zhu Y et al. CKIP-1 recruits nuclear ATM partially to the plasma membrane through interaction with ATM. Cell Signal 2006; 18: 1386–1395.
Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang L et al. EGFR-induced and PKCepsilon monoubiquitylation-dependent NF-kappaB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell 2012; 48: 771–784.
Lee HJ, Lan L, Peng G, Chang WC, Hsu MC, Wang YN et al. Tyrosine 370 phosphorylation of ATM positively regulates DNA damage response. Cell Res 2015; 25: 225–236.
Copley SD . Moonlighting is mainstream: paradigm adjustment required. BioEssays 2012; 34: 578–588.
Sieburth D, Ch'ng Q, Dybbs M, Tavazoie M, Kennedy S, Wang D et al. Systematic analysis of genes required for synapse structure and function. Nature 2005; 436: 510–517.
Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN . Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 2013; 110: 12526–12534.
Dikeakos JD, Atkins KM, Thomas L, Emert-Sedlak L, Byeon IJ, Jung J et al. Small molecule inhibition of HIV-1-induced MHC-I down-regulation identifies a temporally regulated switch in Nef action. Mol Biol Cell 2010; 21: 3279–3292.
Youker RT, Shinde U, Day R, Thomas G . At the crossroads of homoeostasis and disease: roles of the PACS proteins in membrane traffic and apoptosis. Biochem J 2009; 421: 1–15.
Lu WJ, Amatruda JF, Abrams JM . p53 ancestry: gazing through an evolutionary lens. Nature Rev Cancer 2009; 9: 758–762.
Yu J, Zhang L . No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell 2003; 4: 248–249.
Acknowledgements
We thank B Vogelstein, S Narayan, D Chen, G Xiao, C Bakkenist and L Lan for reagents; L Li for assistance with preliminary experiments; and G Xiao and M. Kveiborg for critically reading the manuscript. This work was supported by NIH R01 CA151564 (to GT), HL112791 (to YZ), GM115389 to (JZ), AHA 12SDG9050005 (to JZ) and ALA RG350146 (to JZ).
Author contributions
JB-G, SA, SL, LT, KMA, JEA, JZ, YZ and GT conceived and designed the experiments. JB-G, SA, SL, LT, KMA, LLT and JEA performed the experiments. JB-G, SA, SL, LT, KMA, JEA and YZ analyzed the data. GT wrote the paper. KMA, SA, JEA, JB-G and LLT edited the paper. LT assembled the artwork. All the authors approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Deshmukh
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Barroso-González, J., Auclair, S., Luan, S. et al. PACS-2 mediates the ATM and NF-κB-dependent induction of anti-apoptotic Bcl-xL in response to DNA damage. Cell Death Differ 23, 1448–1457 (2016). https://doi.org/10.1038/cdd.2016.23
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.23
This article is cited by
-
A synergistic partnership between IL-33/ST2 and Wnt pathway through Bcl-xL drives gastric cancer stemness and metastasis
Oncogene (2023)
-
7,8-Dihydroxyflavone improves neuropathological changes in the brain of Tg26 mice, a model for HIV-associated neurocognitive disorder
Scientific Reports (2021)
-
Poly-glycine–alanine exacerbates C9orf72 repeat expansion-mediated DNA damage via sequestration of phosphorylated ATM and loss of nuclear hnRNPA3
Acta Neuropathologica (2020)
-
New factors in mammalian DNA repair—the chromatin connection
Oncogene (2017)
-
Potential Roles of Mitochondria-Associated ER Membranes (MAMs) in Traumatic Brain Injury
Cellular and Molecular Neurobiology (2017)
