
Abstract
Understanding the means by which microglia self-regulate the neuroinflammatory response helps modulating their reaction during neurodegeneration. In amyotrophic lateral sclerosis (ALS), classical NF-κB pathway is related to persistent microglia activation and motor neuron injury; however, mechanisms of negative control of NF-κB activity remain unexplored. One of the major players in the termination of classical NF-κB pathway is the ubiquitin-editing enzyme A20, which has recognized anti-inflammatory functions. Lately, microRNAs are emerging as potent fine-tuners of neuroinflammation and reported to be regulated in ALS, for instance, by purinergic P2X7 receptor activation. In this work, we uncover an interplay between miR-125b and A20 protein in the modulation of classical NF-κB signaling in microglia. In particular, we establish the existence of a pathological circuit in which termination of A20 function by miR-125b strengthens and prolongs the noxious P2X7 receptor-dependent activation of NF-κB in microglia, with deleterious consequences on motor neurons. We prove that, by restoring A20 levels, miR-125b inhibition then sustains motor neuron survival. These results introduce miR-125b as a key mediator of microglia dynamics in ALS.
Similar content being viewed by others


Main
Amyotrophic lateral sclerosis (ALS) is the most widespread and severe form of adult motor neuron (MN) degeneration targeting brainstem, spinal cord and motor cortex. Although the disease is heterogeneous in age and site of onset, as well as rate of progression, the median survival time of patients is usually 3–5 years from diagnosis.1 In most cases, ALS occurs as a sporadic disease, while about 10% of patients suffer from the familial form. Genetic mutations are known to cause familial ALS and the most common are located in the genes encoding superoxide dismutase 1 (SOD1), C9ORF72, fused in sarcoma/translated in liposarcoma (FUS, also known as TLS) and TDP-43 (TARDBP).2 The discovery of these mutations has provided main insights into the pathogenesis of ALS. In particular, the first generated rodent models overexpressing the human SOD1 gene with a substitution of glycine to alanine at position 93 (SOD1-G93A) still provide the best system for translational research, by recapitulating most of human symptoms.3, 4 On the other hand, the recent identification of mutations in genes for TDP-43 and FUS, both involved in RNA splicing, transport, translation and microRNA (miRNA) biogenesis, draws attention to the role of altered RNA and miRNA metabolism in ALS.5, 6
Neuroinflammation dominated by microglia has a central role in MN death and ALS disease progression,7 and mechanisms by which these cells contribute to neuronal damage and degeneration are the subject of intense studying.8, 9 Activation of the classical nuclear factor kappa B (NF-κB) pathway involving the P65/P50 heterodymer is normally transient in microglia, while sustained NF-κB stimulation is associated with persistent neuroinflammation culminating in neuronal tissue damage and death.10 Moreover, P65 is activated in glia from both familial and sporadic ALS cases11 and loss-of-function mutations in the gene encoding optineurin, which negatively regulates tumor necrosis factor α (TNFα)-induced NF-κB activation,12 are found in ALS patients.13 Recently, NF-κB hyper activation has been demonstrated predominately in microglia in the SOD1-G93A mouse, where it regulates the conversion to an inflammatory, neurotoxic phenotype. Consistently, inhibition of NF-κB in microglia extends survival in the ALS mice, by strongly delaying disease progression.14 However, the molecular basis for the pathogenic constitutive activation of NF-κB remains to be explored.
Elaborate negative regulatory mechanisms keep NF-κB signaling in check, in order to maintain tissue homeostasis.15 One of the proteins known to have a key role in the termination of NF-κB signaling is A20 (TNF alpha-induced protein 3, TNFAIP3). A20 is a NF-κB-dependent gene16 that encodes an ubiquitin-editing enzyme essential for the termination of NF-κB activation in response to multiple stimuli, among which are TNFα and lipopolysaccharide (LPS).17, 18, 19, 20 The potent anti-inflammatory properties of A20 are exemplified by the phenotype of the A20 knockout (KO) mice that exhibit persistent NF-κB activation in response to TNFα or sub-lethal doses of LPS, and die prematurely, owing to uncontrolled inflammation.19 In addition, recent studies performed on A20 heterozygous and KO mice brain showed that both total or partial loss of A20 causes spontaneous neuroinflammation characterized by microgliosis, astrogliosis and consequent axonal injury.21
MiRNAs are endogenous small oligonucleotides that bind complementary sequences in mRNAs, usually resulting in gene silencing via translational repression or target degradation.22 In the past years, miRNAs have emerged as key regulators of gene expression, and miRNA dysregulations are associated with many diseases among which is ALS.23 Furthermore, genes mutated in ALS such as TDP-43 and FUS are directly involved in mRNA processing, an additional feature linking miRNAs to ALS.24, 25 MiR-125b is a microglia-enriched miRNA26 that is directly activated by NF-κB27 and shown to cause constitutive NF-κB activation by suppressing A20 expression.28
We previously demonstrated that ALS microglia are hyper reactive to inflammatory stimuli released from injured tissue, such as extracellular ATP acting particularly on pro-inflammatory purinergic P2X7 receptor (P2X7r).29, 30, 31, 32 Moreover, we proved the upregulation of several miRNAs comprising the inflammatory miR-125b in ALS microglia upon stimuli such as TNFα or 2’-3’-O-(benzoyl-benzoyl) ATP (BzATP) acting on P2X7r.33
In this study, we disclose a miR-125b–A20–NF-κB correlation sustaining inflammatory signaling in microglia. Moreover, we demonstrate that repression of A20 by miR-125b is responsible for sustained NF-κB activity in ALS microglia and toxicity towards MNs.
Results
A20 expression is induced in non-transgenic (nt) but not in SOD1-G93A (G93A) microglia upon inflammatory BzATP stimulation
NF-κB-dependent A20 induction upon inflammatory stimuli such as LPS is well known in macrophages,34, 35 poorly documented in CNS microglia36 but never demonstrated upon inflammatory P2X7r stimulation, despite the proven activation of the canonical NF-κB pathway by P2X7r activation in microglia.37
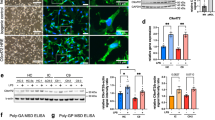
For this reason, we assessed A20 protein expression in both nt and G93A microglia in response to the preferential ligand of P2X7r, BzATP. We used BzATP at the final concentration of 100 μM that is proved not cytotoxic in our system (data not shown). As Figures 1a and b shows, A20 induction is observed after BzATP stimulation of nt cells for 2 h and, moreover, sustained for 24 h (Figures 1d and e). In addition, A20 induction in nt cells is abolished by the specific P2X7r antagonist A839977 (Figures 1d and e). BzATP instead fails to induce A20 protein expression in G93A microglia, at any considered time. In parallel, we established the ratio of ser536-phosphorylated P65 subunit of NF-κB on total P65. Although in G93A microglia the phosphorylation of P65 is increased after BzATP stimulation for 2 h (Figures 1a and c) and sustained for up to 24 h (Figures 1d and f), in nt microglia it is transient and maintained only for 2 h. Consistently, in G93A microglia the phosphorylation of P65 by BzATP for 24 h is prevented by the P2X7r antagonist A839977 (Figures 1d and f). Thus we sought to identify the mechanism(s) by which A20 expression is regulated in ALS microglia. In a previous work, we found that miR-125b is upregulated in primary microglia from SOD1-G93A mice and induced by stimulation for 2 h with BzATP in both nt and G93A microglia.33 As mouse A20 UTR is predicted to interact with miR-125b through a conserved seed sequence (Figure 2a), we hypothesized that miR-125b modulation could be directly responsible for A20 regulation in our system. We assessed the outcome of co-transfecting miR-125b mimic with a construct in which the A20 UTR containing miR-125b target site was cloned downstream of renilla luciferase reporter. By an in vitro assay, we have then proven that miR-125b is able to reduce luciferase activity when co-transfected with A20 UTR, thus demonstrating a regulatory interaction between miR-125b and A20 (Figure 2b). In addition, we have demonstrated that enforced miR-125b overexpression in nt microglia (Figure 2c) is able to reduce A20 endogenous levels (Figure 2d), thus revealing miR-125b as a potential regulator of A20 in microglial cells.

A20 is induced in nt but not in G93A microglia upon inflammatory BzATP stimulation. nt and G93A microglia were exposed to 100 μM BzATP for 2 and 24 h, with or without 1 μM of the P2X7r antagonist A839977. Protein lysates were then subjected to western blotting analysis with the specified antibodies. (a and d) Representative western blotting images and (b and e) statistical analysis of A20 protein expression. (c and f) Western blotting analysis of pP65/P65 ratio. GAPDH antibody was used for normalizations. Normalized values are means±S.E.M. *P≤0.05; **P≤0.02. Statistical significance between two individual groups was assessed using Student’s t-test

A20 is regulated by miR-125b. (a) Schematic representation of miR-125b seed region in A20 3′UTR in mammals (Mmu, mouse; Hsa, human; Ptr, chimpanzee; Mml, rhesus) (b) Normalized luciferase activities of renilla luciferase reporter plasmid with or without A20 3′UTR at 48 h after co-transfection together with miR-125b mimic or scramble mimic in HEK293 cells. (c and d) nt microglia were transfected with miR-125b mimic or scramble mimic. At 48 h after transfection, RNA extracts were subjected to RT-qPCR for miR-125b while protein lysates were subjected to western blotting analysis with A20 antibodies. (e) miR-125b was inhibited in both nt and G93A microglia by transfection of specific hairpin inhibitor. At 48 h after transfection, cells were exposed for 2 h to 100 μM BzATP, and protein lysates were subjected to western blotting analysis with A20 antibodies. GAPDH antibody was used for normalizations. Normalized values are means±S.E.M. *P≤0.05; **P≤0.02. Statistical significance between two individual groups was assessed using Student’s t-test
A20 expression is regulated by miR-125b in microglia
Next, in order to test whether miR-125b is acting within cultured microglia to cause A20 suppression, we inhibited miR-125b by using a specific hairpin inhibitor and assessed A20 protein levels in both nt and G93A microglia upon BzATP stimulation. As shown in Figure 2e, inhibition of miR-125b significantly upregulates A20 protein levels in nt microglia only upon BzATP stimulation and in G93A in both basal and BzATP-stimulated conditions, thus corroborating our hypothesis of regulation of A20 under inflammatory conditions.
MiR-125b expression modulates classical NF-κB/P65 activation upon inflammatory stimuli
Then we exposed nt cells expressing low basal levels of miR-125b to inflammatory BzATP or LPS under conditions of miR-125b overexpression and then assessed NF-κB activation by P65 phosphorylation. The results shown in Figure 3a demonstrate that enforced miR-125b overexpression significantly enhances P65 phosphorylation under all inflammatory conditions tested.

MiR-125b modulates BzATP- or LPS-induced NF-κB/P65 activation. (a) nt microglia were transfected with miR-125b mimic or scramble mimic. At 48 h after transfection, cells were treated with 100 μM BzATP or LPS 100 ng/ml for 2 h and protein lysates were subjected to western blotting analysis with the specified antibodies. (b and c) miR-125b was inhibited in nt and/or G93A microglia by transfection of specific hairpin inhibitor. At 48 h after transfection, cells were exposed for 2 h to 100 μM BzATP (b) or LPS 100 ng/ml (c), and protein lysates were subjected to western blotting analysis with antibodies for total P65 (P65) and phospho-P65 (pP65). GAPDH antibody was used for normalizations. Normalized values are means±S.E.M. *P≤0.05; **P≤0.02. Statistical significance between two individual groups was assessed using Student’s t-test
Conversely, to test whether miR-125b inhibition could reduce canonical NF-κB activity, we measured P65 phosphorylation following BzATP exposure in nt and G93A microglia in the presence of miR-125b inhibitor. Even though P65 phosphorylation is significantly further enhanced by BzATP in G93A microglia when compared with nt cells, this assay demonstrated that miR-125b inhibition is able to downmodulate BzATP-induced P65 phosphorylation in both nt and G93A (Figure 3b). The same occurred in the presence of LPS (Figure 3c), thus extending the role of miR-125b as a modulator of classical NF-κB inflammatory activators. We also observed a trend of total P65 downmodulation in the presence of miR-125b inhibition that, however, did not reach statistical significance (data not shown).
Classical NF-κB/P65 regulation by miR-125b is dependent on A20
A20 functions to terminate classical NF-κB activation following cellular exposure to various pathogens and proinflammatory cytokines. These activities are well characterized upon several stimuli, including TNFα and LPS.38, 20 Assuming that miR-125b upregulation, by regulating A20 levels as demonstrated in B cells,32 could affect classical NF-κB activation also in microglia, we next determined whether the reduction of P65 phosphorylation through miR-125b inhibition was dependent on its target A20. To reach this aim, A20 was silenced by transfection with a pool of specific siRNAs for 48 h. The efficiency of silencing was determined by western blotting on total protein lysates, showing that A20 protein expression was knocked down by >70% (Figure 4a).

Classical NF-κB/P65 phosphorylation is regulated by miR-125b through A20. (a) A20 expression in total protein lysates is measured by western blotting analysis after A20 silencing in G93A microglia, in the presence or absence of miR-125b inhibition. (b) NF-κB/P65 phosphorylation was measured in total protein lysates by P65 protein phosphorylation in G93A microglia stimulated with BzATP for 2 h, in the presence of miR-125b inhibition and with or without silencing of A20. Normalized values are means±S.E.M. *P≤0.05; **P≤0.02. Statistical significance between two individual groups was assessed using Student’s t-test
As depicted in Figure 4b, silencing of A20 in the presence of miR-125b inhibition completely restored BzATP-mediated P65 phosphorylation, thus establishing A20 as a mediator of miR-125b effect on classical NF-κB pathway.
MiR-125b inhibition reduces the expression of transcriptional targets of NF-κB
In a previous work, we showed that TNFα transcription is enhanced in G93A with respect to nt microglia, and this effect is dependent on miR-125b expression.33 As TNFα is a well-known NF-κB target gene39 that is moreover induced by P2X7r activation in microglia,33, 40 we decided to extend the potential effects of miR-125b inhibition to TNFα mRNA levels in both nt and G93A microglia. By performing RT-qPCR, we found that the increase in TNFα levels by BzATP is strongly prevented in both nt and G93A microglia by miR-125b inhibition (Figure 5a).

MiR-125b inhibition regulates TNFα and NOX2. MiR-125b was inhibited in both nt and G93A microglia by transfection of specific hairpin inhibitor. At 48 h after transfection, cells were exposed for 2 h to 100 μM BzATP. RNA extracts were analyzed by RT-qPCR with specific primers for mouse (a) TNFα or (b) NOX2 using ΔΔCt method, while protein extracts were subjected to western blotting analysis with (c) gp91phox antibody. (d) MiR-125b was inhibited in both nt and G93A microglia by transfection of a specific hairpin inhibitor. Protein extracts were subjected to western blotting analysis with P2X7r antibody. GAPDH antibody was used for normalizations. Normalized values are means±S.E.M. *P≤0.05; **P≤0.02. Statistical significance between two individual groups was assessed using Student’s t-test
As for TNFα, NOX2 gene encoding for gp91phox is also a proven transcriptional NF-κB target in microglia.41 Despite a mild stimulation of mRNA level by BzATP in both nt and G93A microglia, we demonstrated that miR-125b inhibition was also able to restore NOX2 mRNA levels to basal unstimulated conditions only in G93A microglia (Figure 5b). Similarly, as Figure 5c shows, after inflammatory BzATP challenge of G93A microglia, we have found a significant upregulation of gp91phox at the protein level that was also abrogated by miR-125b inhibition. In order to test whether the inhibitory effects of anti-125b could be attributable to a variation in P2X7r levels, we analyzed total receptor protein in both nt and G93A microglia after inhibiting miR-125b for 48 h. As reported in Figure 5d, P2X7r levels remain constant.
The NADPH oxidase 2 (NOX2) complex is formed by gp91phox, the product of the NOX2 gene, which constitutively resides on the plasma membrane, and cytosolic factors among which is P67phox, the product of the Ncf2 gene, which translocates to the membrane in response to cellular stimuli to activate gp91phox.42 In a previous work, we have demonstrated that G93A microglia shows an enhanced translocation of P67phox upon inflammatory BzATP stimulation when compared with nt cells.29 Here we asked whether miR-125b inhibition, by acting on NF-κB/P65 pathway, could also affect the membrane-bound P67phox levels in G93A microglia upon BzATP stimulation. We found that miR-125b inhibition exerts an inhibitory effect on P67phox translocation, moreover comparable to that obtained with the P2X7r antagonist Brilliant Blue G (Figure 6a).

MiR-125b inhibition regulates NOX2 activation and ROS production. (a) miR-125b was inhibited in G93A microglia by transfection of specific hairpin inhibitor. At 48 h after transfection, cells were exposed for 2 h to 100 μM BzATP with or without 1μM Brilliant Blue G (BBG), and equal amount of proteins from isolated plasma membranes were subjected to western blotting and immunoreactions with the specified antibodies. (b) miR-125b was inhibited in G93A microglia by transfection of specific hairpin inhibitor. At 48 h after transfection, cells were exposed for 2 h to 100 μM BzATP with or without 1 μM BBG and then exposed to H2DCF-DA. Fluorescence was visualized using fluorescence microscopy (scale bar=50 μM). Total cell numbers and green fluorescent cells were counted in the visual field, and the percentage of ROS/DCF-DA-positive cells was calculated. Normalized values are means±S.E.M. *P≤0.05, **P≤0.02 versus untreated nt; #P≤0.05 versus BzATP treated nt. Statistical significance between two individual groups was assessed using Student’s t-test
As the activation of NOX2 by BzATP results in increased generation of reactive oxygen species (ROS),29 we investigated the role of miR-125b inhibition on this same parameter. As shown in Figure 6b, BzATP treatment significantly increases the number of cells incorporating fluorescent 2’,7’-dichlorofluorescein (DCF), a marker of ROS production. Interestingly, miR-125b inhibition prevents the intracellular accumulation of DCF, thus indicating that ROS production via inflammatory P2X7r activation is a downstream target of miR-125b action.
MiR-125b inhibition through A20 protein protects MNs from death induced by activated G93A microglia
To determine whether the inhibitory effect on inflammatory properties of G93A microglia by miR-125b inhibition could turn into a positive action on MN viability, we analyzed microglial-mediated neuronal injury using a conditioned medium (CM) assay. MiR-125b was inhibited in both nt and G93A microglia, and the cells were stimulated with BzATP or LPS for 24 h. We then tested the effects of microglia CM on primary MN-enriched cultures.
As expected from previous results obtained with NSC-34-hSOD1-G93A and SH-SY5Y-hSOD1-G93A cell lines,30 plain medium incubated for 24 h at 37 °C with BzATP or LPS and then added to primary MN-enriched cultures did not cause per se MN death (data not shown). Conversely, primary MN-enriched cultures incubated with CM from G93A microglia challenged with BzATP showed statistically significant MN death. Instead, CM from both nt and G93A microglia challenged with LPS caused MN death in primary enriched cultures (Figure 7a). Most importantly, increased MN survival was always obtained following miR-125b inhibition, as shown by immunofluorescence analysis (Figures 7a and b) and confirmed by western blotting (Figure 7c). However, in the absence of A20, CM from G93A microglia activated with BzATP or lps reverted the protective effect of miR-125b inhibition on MN (Figures 7a–c).

MiR-125b inhibition protects MNs from death induced by activated G93A microglia CM. miR-125b was inhibited in the presence or absence of A20 siRNA in both nt and G93A microglia stimulated with BzATP or LPS for 24 h. The microglia CM was collected and applied for 48 h to primary MN-enriched cultures. (a, b) MN-enriched cultures were then subjected to quantification of MN after immunofluorescence and confocal analysis or (c) to western blotting with SMI32. Normalized values are means±S.E.M. *P≤0.05; ***P≤0.001. Scale bar: 50 μm. Statistical significance between two individual groups was assessed using Student’s t-test
MiR-125b and A20 protein expression levels are inversely modulated in lumbar spinal cord of SOD1-G93A mice at end stage
As microglia activation is a hallmark of diseased spinal cord in ALS, we directly analyzed miR-125b and A20 protein expression in lumbar spinal cord of SOD1-G93A mice at end stage (~23 weeks). Quantitative RT-PCR (RT-qPCR) identified that miR-125b (Figure 8a), similarly to NOX2 mRNA (Figure 8b), is significantly upregulated compared with that of nt mice, whereas the protein level of A20 is strongly inhibited, as demonstrated by western blotting analysis (Figure 8c).

MiR-125b and A20 expression levels are inversely modulated in SOD1-G93A mice lumbar spinal cords at end stage. Total RNA from C57BL/6 J mice and SOD1-G93A mice was subjected to RT-qPCR for (a) miR-125b and (b) NOX2 quantification by ΔΔCt method. U6 and GAPDH were used, respectively, for normalization. (c) Protein lysates from C57BL/6 J mice and SOD1-G93A mice were subjected to western blotting analysis with specified antibodies. Normalized values are means±S.E.M. *P≤0.05; **P≤0.02. Statistical significance between two individual groups was assessed using Student’s t-test
Discussion
It is widely accepted that MN degeneration and ALS progression are a result of a complex interplay between multiple pathogenic mechanisms involving not only the motoneuron itself but also its non-neuronal neighbors, particularly glial cells.43 Astrocytes are the largest glial cellular population in the CNS and reactive astrocytosis, associated with insufficient production of neurotrophic factors and excitotoxicity caused by impaired glutamate clearance, is strongly implicated in MN death.44 Moreover, astrocytosis in ALS also contributes to altered immune response and consequent neuroinflammation, which is one of the pathophysiological mechanisms contributing to MN degeneration in ALS.45, 46 The immune-competent cells of CNS, microglia, also have a dynamic and complex role in the neuroinflammatory component of ALS. After an initial triggering of microglia through factors mainly released by damaged MN and astrocytes, the cell activation process can next be either counteracted or exacerbated, with a final outcome consequently turned from beneficial to toxic. Among the inflammatory elements acting on microglia, purinergic signaling sustained by extracellular ATP mainly through P2X7r,47 and miRNAs as fine-tuners of posttranscriptional regulation, are just emerging as strategic effectors of the inflammatory pathway in ALS.23, 48 Indeed, inhibition of pro-inflammatory miR-155 in SOD1 mice ameliorates disease pathology, strongly suggesting miRNAs as therapeutic targets for the treatment of ALS,49 and P2X7r is lately emerging as gene modifier in ALS.29, 30, 31, 32 In this regard, in the previous work we have characterized the miRNA signature of ALS microglia in both resting and P2X7r-activated conditions, by detecting a specific subset of dysregulated inflammatory miRNAs that might contribute to ALS alterations.33 In particular, we have shown that miR-125b expression is increased in microglia from ALS mice and even further induced in response to inflammatory stimuli such as BzATP acting on P2X7r.33 In addition, two ALS causative genes, TDP43 and FUS, are involved particularly in miR-125b processing, thus contributing to associate this specific miRNA to ALS disease.25, 24
The aim of this study was to further extend our findings on the role of miR-125b as a crossover in P2X7r-mediated neuroinflammation and, in particular, to study whether and how miR-125b can eventually tip the balance between protective and deleterious neuroinflammation in ALS.
Recent findings have demonstrated a mutual activation circuit subsisting in B cells between miR-125b and NF-κB.28 Moreover, activation of P2X7r leads to increased expression of miR-125b in microglia,33 as well as to activation of NF-κB.37, 50 Here we have demonstrated that inhibition of miR-125b is able to decrease both BzATP- and LPS-induced activation of NF-κB in G93A microglia, thus confirming that the cross-regulation between miR-125b and NF-κB occurs also in ALS and, moreover, reinforcing the pro-inflammatory action of miR-125b previously demonstrated in macrophages.51 Our results are also in line with studies performed on Alzheimer’s disease models, where miR-125b was shown to be involved in NF-κB-dependent inflammatory and oxidative stress pathways.52
Activation of NF-κB signaling exerts its pro-inflammatory function in microglia by activating the transcription of genes responsible for the so called microglia M1 activation state.41, 53, 54 Among these, TNFα and NOX2 are well characterized as ALS neuroinflammatory markers.55 In line with the repressive effect exerted on NF-κB by miR-125b inhibition, we observed a downmodulation of TNFα expression in both nt and ALS microglia after inflammatory stimulation of P2X7r. Because, differently from TNFα, we have shown a repression of NOX2 transcription by miR-125b inhibition only in SOD1-G93A, but not in control microglia, the axis miR-125b/NF-κB demonstrated for TNFα might instead act on the NOX2 gene promoter only during the ALS pathological context.
The toxicity of ALS microglia toward MNs is dependent on classical NF-κB activation in vivo14 and in vitro following LPS stimulation.56 Moreover, sustained NF-κB stimulation is associated with persistent neuroinflammation culminating in neuronal damage and death.10 We have characterized here that the tight control of miR-125b expression in ALS primary microglia that is fundamental for the progression into the NF-κB/TNFα/NOX2/ROS pathway induced by P2X7r activation indeed mediates also a noxious inflammatory response with consequent direct impact on MN injury. All these deleterious pathways culminating in MN cell death are in fact totally abrogated by direct inhibition of miR-125b, as we have indeed shown.
The protein A20 is a validated target of miR-125b in humans28 and the seed sequence is strongly conserved among mammals. Having demonstrated that A20 is a posttranscriptional target of miR-125b in mouse microglia and that the A20 protein level is inversely correlated with the miR-125b expression in G93A microglia, we have next hypothesized that regulation of the microglia miR-125b/NF-κB/MN injury axis can be obtained through a modulated expression of the protein A20 in G93A microglia. We have shown here that the silencing of A20 is indeed able to reactivate NF-κB following BzATP treatment in G93A microglia in the simultaneous and deactivating presence of miR-125 inhibitor and is moreover able to switch back the MNs from survival to death. Not surprisingly, we have also found that A20 protein expression is drastically reduced in lumbar spinal cord of ALS mice at end stage, when MN loss is prominent, and is reduced in primary G93A microglia compared with non-transgenic cells, after inflammatory stimulation of P2X7r by BzATP.
From all these results, A20 therefore emerges as a crucial 'node' in the complex network of molecular interactions that sustain the ALS phenotype both in vitro and in vivo and as a novel effector in the complex purinergic signaling taking place in ALS. Because A20 is a feedback-loop suppressor of NF-κB signaling to halt the inflammatory response20, 57of several stimuli such as TNFα and LPS,20, 58, 59 and A20 deficiency has been shown to cause spontaneous neuroinflammation,21 the absence of A20 production in G93A microglia activated by BzATP might thus be one of the mechanisms leading to the persistent microglia activation that occurs in ALS.7
In conclusion, our work has dissected the causative role of miR-125b and its target protein A20 in determining the progression of an inflammatory insult, in particular the one evoked by activation of P2X7r, into SOD1-G93A MN death (Figure 9). Although being aware that the miR-125b/A20 node might only be one of the molecular circuits activated in ALS, it might surely be effective to tip the balance between good and bad neuroinflammation not only in ALS but also in other inflammatory pathologies involving chronic dysregulation of NF-κB signaling.

Proposed mechanism by which miR-125b regulates ALS microglia phenotype and toxicity. In microglia, classical NF-κB/P65 pathway is activated by inflammatory stimuli such as ATP acting on P2X7r or LPS acting on Toll-like receptor (TLR). Once activated, P65 allows transcription of several inflammatory genes (TNFα, Ncf2, NOX2) together with NF-κB negative regulators, among which A20 is necessary to correctly stop the inflammatory response. In ALS microglia, miR-125b is upregulated in response to inflammatory stimuli and negatively regulates A20 protein production, resulting in persistent NF-κB activation and release of toxic factors affecting MN survival
Materials and methods
Reagents
BzATP, LPS and all reagents were obtained from Sigma-Aldrich (Milan, Italy), unless otherwise stated. A-839977 was from Tocris Bioscience (Bristol, UK).
Mice
Adult B6.Cg-Tg(SOD1-G93A)1Gur/J mice expressing high copy number of mutant human SOD1 with a Gly93Ala substitution (SOD1-G93A) were originally obtained from Jackson Laboratories (Bar Harbor, ME, USA) and bred in indoor animal facility.
All animal procedures have been performed according to the European Guidelines for the use of animals in research (86/609/CEE) and the requirements of Italian laws (D.L. 116/92). Ethical procedures have been approved by the Animal Welfare Office, Department of Public Health and Veterinary, Nutrition and Food Safety, General Management of Animal Care and Veterinary Drugs of the Italian Ministry of Health. All efforts were made to minimize animal suffering and to limit the number of animals necessary to produce reliable results. Transgenic progeny was genotyped analyzing tissue extracts from tail tips as described in Apolloni et al.31
Primary microglia cultures
Mixed glial cultures from brain cortex were prepared as previously described.30 Briefly, neonatal SOD1-G93A and non-transgenic littermate mice were killed, and after removing the meninges, cortices were minced and digested with 0.01% trypsin and 10 μg/ml DNaseI. After dissociation and passage through 70-μm filters, cells were suspended in DMEM/F-12 media with GlutaMAX (Life Technologies, Carlsbad, CA, USA), plus 10% fetal bovine serum (FBS), 100 μg/ml gentamicine and 100 U/ml streptomycin/penicillin at a density of about 60 × 103 cells/cm2. After 15 days, a mild trypsinization (0.08% in DMEM/F-12 without FBS) was performed for 40 min at 37 °C to remove non-microglial cells. The resultant adherent microglial cells (pure >98%) were washed twice with DMEM/F-12 and kept in 50% mixed glial cells CM at 37 °C in a 5% CO2 and 95% air for 48 h until used.
Spinal cord MN-enriched cultures and immunofluorescence studies
Spinal cord MN-enriched cultures were prepared from 15-day-old C57BL/6 J embryos.60 Each neural tube was dissected, singularly incubated for 10 min in 0.025% trypsin, transferred into a 0.1 mg/ml DNase1 solution and gently dissociated. The resulting mixed cultures were seeded on poly-d-lysine-coated glass cover slips (about 70 × 104 cells for each glass cover slip) and maintained in neurobasal medium supplemented with B-27 supplement, 0.5 mM glutamine, 5% FBS and 5% horse serum. Cultures were maintained at a 37 °C humidified incubator in 5% CO2 atmosphere. To suppress glia proliferation, at 3 days after seeding Ara-C was added to a final concentration of 10 μM. Cells were cultured for up to 7 days before being used. Following the in vitro treatments, cultures were fixed (4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4) and stained with SMI-32 (Covance, Emeryville, CA, USA; 1 : 500) to visualize MNs. Immunofluorescence was examined by confocal laser scanning microscope (LSM700 Zeiss, Oberkochen, Germany) equipped with four laser lines: 405, 488, 561 and 639 nm. The brightness and contrast of the digital images were adjusted using the Zen software (Zeiss). The criteria adopted for identifying MNs were: immunoreactivity of the cell body for SMI32 (>25 μm in diameter); and extension of axon and dendrites on a multipolar structure. Five independent cultures per treatment group were used.
Cell transfection
Primary microglia (5 × 105/well) were plated and transfection of 20 nM miRIDIAN Hairpin inhibitors, mimic or smart pool siRNA (Dharmacon Products, Thermo Fisher Scientific, Rockford, IL, USA) was performed with lipofectamine 2000 (Invitrogen, Life Technologies) for 48 h, according to the manufacturers' instructions. Specifically, in order to silence A20 we used siGENOME SMART POOL mouse TNFAIP3, a mix of three different siRNA designed on A20 coding region, and siGENOME Control pool non-targeting, as negative control (Dharmacon Products, Thermo Fisher Scientific). All mimic, inhibitor and siRNA negative controls are based on Caenorhabditis elegans cel-miR-67 sequence not present in humans (Dharmacon Products, Thermo Fisher Scientific).
Protein extraction, SDS-PAGE and western blotting
Protein lysates were obtained by homogenization of mice lumbar spinal cord segments in homogenization buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 1% Triton X-100, 10 mM EDTA) added with protease inhibitor cocktail (Sigma-Aldrich). After sonication, lysates were kept for 30 min on ice and then centrifuged for 20 min at 14 000 × g at 4 °C. Cells in serum-free medium were harvested with ice-cold RIPA buffer (PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS), added with protease inhibitor cocktail (Sigma-Aldrich). Lysates were kept for 30 min on ice and then centrifuged for 10 min at 14 000 × g at 4 °C. Supernatants were collected and assayed for protein quantification by the BCA method (Thermo Fischer Scientific). Analysis of protein components was performed by polyacrylamide gel electrophoresis separation (Bio-Rad, Milan, Italy) and transferred onto nitrocellulose membranes (Amersham Biosciences, Cologno Monzese, Italy). After saturation, blots were probed overnight at 4 °C, with the specified antibody, and finally incubated for 1 h with HRP-conjugated secondary antibodies and detected on X-ray film (Aurogene, Rome, Italy) using the ECL Advance Western Blotting Detection Kit (Amersham Biosciences). Quantifications were performed by Kodak Image Station (Rochester, NY, USA). In all cases, GAPDH was used for protein normalization.
Membrane translocation of P67phox
After treatment, primary microglia were lysed in relaxation buffer (100 mM KCl, 3 mM NaCl, 3.5 mM MgCl2, 1.25 mM EGTA and 10 mM Pipes, pH 7.3) added with protease inhibitor cocktail (Sigma-Aldrich) and sonicated (3 × 10 s, 4 °C using a microprobe sonicator). Cell lysate was centrifuged at 500 × g to remove the heavy mitochondria and nuclei and to generate a postnuclear supernatant (PNS). The PNS was subsequently ultracentrifuged at 100 000 × g for 1 h a 4 °C to spin down total membranes. The membranes were suspended in 100 μl of relaxation buffer added with 1% Triton X-100; protein concentration was estimated using BCA assay and 5 μg of protein were subjected to SDS-PAGE and probed with anti-P67phox and gp91phox antibodies.
Antibodies
The following antibodies were used and obtained as follows: A20 mouse monoclonal antibody (1 : 500) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); NF-κB P65 rabbit polyclonal antibody (1 : 1000) from Cell Signalling Technology Inc. (Beverly, MA, USA); phospho-NF-κB P65 (Ser536) rabbit polyclonal antibody (1 : 1000) from Cell Signalling Technology Inc.; gp91 phox mouse monoclonal antibody (1 : 1000) from BD Transduction Laboratories (Lexington, KY, USA); P67 mouse monoclonal antibody (1 : 1000) BD Transduction Laboratories; SMI32 mouse monoclonal antibody (1 : 1000) from Covance; CD68 rabbit polyclonal antibody (1 : 1000) from Santa Cruz Biotechnology; P2X7r rabbit polyclonal antibody (1 : 500) from Alomone Labs (Jerusalem, Israel); and GAPDH mouse monoclonal antibody (1 : 10000) from Calbiochem (San Diego, CA, USA).
Intracellular ROS measurement
Intracellular ROS production was monitored by the permeable fluorescent dye, 2' 7'-dichlorodihydrofluorescein diacetate acetyl ester (H2DCF-DA; Molecular Probes, Carlsbad, CA, USA), which permeates into the cells and is oxidized in the presence of ROS. After the indicated treatment, cells were incubated with Krebs-Ringer HEPES buffer (1.3 mM CaCl2, 131 mM NaCl, 1.3 mM MgSO4, 5 mM KCl, 0.4 mM KH2PO4, 6 mM Glucose, 20 mM HEPES) containing 10 μM of H2DCF-DA for 60 min at 37 °C and washed three times with PBS. Fluorescence was then analyzed using a fluorescence microscope (JuLI, Digital Bio, Rockester, NY, USA). The images were captured with CMOS 1.3 M pixel camera (Rockester, NY, USA) and exported as TIFF files. Total cell numbers in the visual field and the green fluorescent cells were counted, and the percentage of ROS/H2DCF-DA-positive cells was calculated.
Quantitative RT-PCR
Total RNA, including small RNA, was extracted with TRIZOL (Invitrogen, Life Technologies) according to the manufacturer’s instruction and was checked with the Nanodrop 100 System (Rockford, IL, USA) and the Agilent 2100 bioanalyzer (Santa Clara, CA, USA). QPCR for miRNA quantification was conducted using the miScript SYBR Green PCR Kit (Qiagen, Hilden, Germany) subsequent to reverse transcription using the miScript Reverse Transcription Kit (Qiagen) according to the manufacturer's instructions. All miRNA primers were purchased from Qiagen, and the relative expression levels were calculated using ΔΔCt method with mouse U6 small nuclear RNA as the normalizing control. QPCR for TNFα and NOX2 quantification was performed using SYBR Green select (Applied Biosystems, Milan, Italy) subsequent to reverse transcription using the Superscript Vilo cDNA Synthesis Kit (Life Technologies). Relative gene expression was calculated by ΔΔCt analysis relative to GAPDH expression levels. Primers were as follows: TNFα forward 5′-CTGTAGCCCACGTCGTAGC-3′; TNFα reverse 5′-TTGAGATCCATGCCGTTG-3′; GAPDH forward 5′-CATGGCCTTCCGTGTTCCTA-3′; and GAPDH reverse 5′-CCTGCTTCACCACCTTCTTGAT-3′.
Luciferase assay
The 3′UTR containing miR-125b seed region of mouse A20 was PCR amplified with XbaI flanked primers (F: 5′-gcTCTAGAGTGCGAACACATTGACAG-3′; R: 5′-gcAGATCTCTAGAAGGTGCATACAGC-3′). The PCR products were purified, digested and cloned downstream of the luciferase coding region in the pRL-TK vector (Promega, Milan, Italy). Human HEK293T cell line was co-transfected with control mimic or miR-125b mimic (Dharmacon Products, Thermo Fisher Scientific) and pRL-TK vector containing A20 UTR along with firefly luciferase expression plasmid (PGL3) as a transfection control using Lipofectamine (Invitrogen, Life Technologies). After 48 h, cells were lysed and analyzed for relative luciferase activity using the Dual Luciferase Assay Kit (Promega). Results are representative of at least three independent experiments.
Statistical analysis
Data are presented as mean±S.E.M. Normality of data was assessed by Shapiro–Wilk test. Statistical differences were verified by Student’s unpaired two-tailed t-test using MedCalc (Medcalc Software, Mariakerke, Belgium). *P<0.05 was considered significant.
Abbreviations
- ALS:
-
amyotrophic lateral sclerosis
- BzATP:
-
2’-3’-O-(benzoyl-benzoyl) ATP
- CM:
-
conditioned medium
- MN:
-
motor neuron
- NF-κB:
-
nuclear factor kappa B
- LPS:
-
lypopolysaccaride
- miRNA:
-
microRNA
- P2X7r:
-
P2X7 receptor
- NOX2:
-
NADPH oxidase 2
- nt:
-
non transgenic
- SOD1:
-
superoxide dismutase 1
- G93A:
-
SOD1-G93A
- TNFα:
-
tumor necrosis factor alpha
References
Swinnen B, Robberecht W . The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 2014; 10: 661–670.
Renton AE, Chio A, Traynor BJ . State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014; 17: 17–23.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994; 264: 1772–1775.
Poppe L, Rue L, Robberecht W, Van Den Bosch L . Translating biological findings into new treatment strategies for amyotrophic lateral sclerosis (ALS). Exp Neurol 2014; 262: 138–151.
Robberecht W, Philips T . The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013; 14: 248–264.
Paez-Colasante X, Figueroa-Romero C, Sakowski SA, Goutman SA, Feldman EL . Amyotrophic lateral sclerosis: mechanisms and therapeutics in the epigenomic era. Nat Rev Neurol 2015; 11: 266–279.
Philips T, Rothstein JD . Glial cells in amyotrophic lateral sclerosis. Exp Neurol 2014; 262: 111–120.
Brites D, Vaz AR . Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front Cell Neurosci 2014; 8: 117.
D'Ambrosi N, Rossi S, Gerbino V, Cozzolino M . Rac1 at the crossroad of actin dynamics and neuroinflammation in amyotrophic lateral sclerosis. Front Cell Neurosci 2014; 8: 279.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH . Mechanisms underlying inflammation in neurodegeneration. Cell 2010; 140: 918–934.
Swarup V, Phaneuf D, Dupre N, Petri S, Strong M, Kriz J et al. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med 2011; 208: 2429–2447.
Zhu G, Wu CJ, Zhao Y, Ashwell JD . Optineurin negatively regulates TNFalpha- induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol 2007; 17: 1438–1443.
Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010; 465: 223–226.
Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 2014; 81: 1009–1023.
Renner F, Schmitz ML . Autoregulatory feedback loops terminating the NF-kappaB response. Trends Biochem Sci 2009; 34: 128–135.
Opipari AW Jr, Boguski MS, Dixit VM . The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem 1990; 265: 14705–14708.
Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004; 430: 694–699.
Vereecke L, Beyaert R, van Loo G . The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 2009; 30: 383–391.
Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000; 289: 2350–2354.
Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 2004; 5: 1052–1060.
Guedes RP, Csizmadia E, Moll HP, Ma A, Ferran C, da Silva CG . A20 deficiency causes spontaneous neuroinflammation in mice. J Neuroinflammation 2014; 11: 122.
Bartel DP . MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–233.
Volonté C, Apolloni S, Parisi C . MicroRNAs: newcomers into the ALS picture. CNS Neurol Disord Drug Targets 2015; 14: 194–207.
Morlando M, Dini Modigliani S, Torrelli G, Rosa A, Di Carlo V, Caffarelli E et al. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J 2012; 31: 4502–4510.
Di Carlo V, Grossi E, Laneve P, Morlando M, Dini Modigliani S, Ballarino M et al. TDP-43 regulates the microprocessor complex activity during in vitro neuronal differentiation. Mol Neurobiol 2013; 48: 952–963.
Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 2014; 17: 131–143.
Ma X, Becker Buscaglia LE, Barker JR, Li Y . MicroRNAs in NF-kappaB signaling. J Mol Cell Biol 2011; 3: 159–166.
Kim SW, Ramasamy K, Bouamar H, Lin AP, Jiang D, Aguiar RC . MicroRNAs miR-125a and miR-125b constitutively activate the NF-kappaB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc Natl Acad Sci USA 2012; 109: 7865–7870.
Apolloni S, Parisi C, Pesaresi MG, Rossi S, Carri MT, Cozzolino M et al. The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J Immunol 2013; 190: 5187–5195.
D'Ambrosi N, Finocchi P, Apolloni S, Cozzolino M, Ferri A, Padovano V et al. The proinflammatory action of microglial P2 receptors is enhanced in SOD1 models for amyotrophic lateral sclerosis. J Immunol 2009; 183: 4648–4656.
Apolloni S, Amadio S, Montilli C, Volonté C, D'Ambrosi N . Ablation of P2X7 receptor exacerbates gliosis and motoneuron death in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet 2013; 22: 4102–4116.
Apolloni S, Amadio S, Parisi C, Matteucci A, Potenza RL, Armida M et al. Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis Model Mech 2014; 7: 1101–1109.
Parisi C, Arisi I, D'Ambrosi N, Storti AE, Brandi R, D'Onofrio M et al. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis 2013; 4: e959.
Liu B, Jiang D, Ou Y, Hu Z, Jiang J, Lei X . An anti-inflammatory role of A20 zinc finger protein during trauma combined with endotoxin challenge. J Surg Res 2013; 185: 717–725.
Lai TY, Wu SD, Tsai MH, Chuang EY, Chuang LL, Hsu LC et al. Transcription of Tnfaip3 is regulated by NF-kappaB and p38 via C/EBPbeta in activated macrophages. PLoS One 2013; 8: e73153.
Austin JW, Gilchrist C, Fehlings MG . High molecular weight hyaluronan reduces lipopolysaccharide mediated microglial activation. J Neurochem 2012; 122: 344–355.
Ferrari D, Wesselborg S, Bauer MK, Schulze-Osthoff K . Extracellular ATP activates transcription factor NF-kappaB through the P2Z purinoreceptor by selectively targeting NF-kappaB p65. J Cell Biol 1997; 139: 1635–1643.
Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014; 512: 69–73.
Collart MA, Baeuerle P, Vassalli P . Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol 1990; 10: 1498–1506.
Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K et al. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem 2000; 75: 965–972.
Lim H, Kim D, Lee SJ . Toll-like receptor 2 mediates peripheral nerve injury-induced NADPH oxidase 2 expression in spinal cord microglia. J Biol Chem 2013; 288: 7572–7579.
Leto TL, Morand S, Hurt D, Ueyama T . Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal 2009; 11: 2607–2619.
Mancuso R, Navarro X . Amyotrophic lateral sclerosis: current perspectives from basic research to the clinic. Prog Neurobiol 2015; 133: 1–26.
Valori CF, Brambilla L, Martorana F, Rossi D . The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell Mol Life Sci 2014; 71: 287–297.
Endo F, Komine O, Fujimori-Tonou N, Katsuno M, Jin S, Watanabe S et al. Astrocyte-derived TGF-beta1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep 2015; 11: 592–604.
Zhao W, Beers DR, Appel SH . Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol 2013; 8: 888–899.
Volonté C, Apolloni S, Skaper SD, Burnstock G . P2X7 receptors: channels, pores and more. CNS Neurol Disord Drug Targets 2012; 11: 705–721.
Volonté C, Apolloni S, Carri MT, D'Ambrosi N . ALS: focus on purinergic signalling. Pharmacol Ther 2011; 132: 111–122.
Butovsky O, Jedrychowski MP, Cialic R, Krasemann S, Murugaiyan G, Fanek Z et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol 2015; 77: 75–99.
Aga M, Watters JJ, Pfeiffer ZA, Wiepz GJ, Sommer JA, Bertics PJ . Evidence for nucleotide receptor modulation of cross talk between MAP kinase and NF-kappa B signaling pathways in murine RAW 264.7 macrophages. Am J Physiol Cell Physiol 2004; 286: C923–C930.
Chaudhuri AA, So AY, Sinha N, Gibson WS, Taganov KD, O'Connell RM et al. MicroRNA-125b potentiates macrophage activation. J Immunol 2011; 187: 5062–5068.
Zhao Y, Bhattacharjee S, Jones BM, Hill J, Dua P, Lukiw WJ . Regulation of neurotropic signaling by the inducible, NF-kB-sensitive miRNA-125b in Alzheimer's disease (AD) and in primary human neuronal-glial (HNG) cells. Mol Neurobiol 2014; 50: 97–106.
Colton CA . Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 2009; 4: 399–418.
Ivashkiv LB . Inflammatory signaling in macrophages: transitions from acute to tolerant and alternative activation states. Eur J Immunol 2008; 41: 2477–2481.
Beers DR, Zhao W, Liao B, Kano O, Wang J, Huang A et al. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav Immun 2011; 25: 1025–1035.
Valente T, Mancera P, Tusell JM, Serratosa J, Saura J . C/EBPbeta expression in activated microglia in amyotrophic lateral sclerosis. Neurobiol Aging 2012; 33: 2186–2199.
Catrysse L, Vereecke L, Beyaert R, van Loo G . A20 in inflammation and autoimmunity. Trends Immunol 2014; 35: 22–31.
Coornaert B, Carpentier I, Beyaert R . A20: central gatekeeper in inflammation and immunity. J Biol Chem 2009; 284: 8217–8221.
Turer EE, Tavares RM, Mortier E, Hitotsumatsu O, Advincula R, Lee B et al. Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J Exp Med 2008; 205: 451–464.
Spalloni A, Albo F, Ferrari F, Mercuri N, Bernardi G, Zona C et al. Cu/Zn-superoxide dismutase (GLY93—>ALA) mutation alters AMPA receptor subunit expression and function and potentiates kainate-mediated toxicity in motor neurons in culture. Neurobiol Dis 2004; 15: 340–350.
Acknowledgements
We thank Dr. Cristiana Valle for useful suggestions and advice with cloning and Professor Maria Teresa Carrì for critical reading of the manuscript. This study was supported by Italian Ministry for Education, University and Research in the framework of the Flagship Project NanoMAX to CV and by AriSLA grant MAMMALS to CP.
Author contributions
CP designed, performed the experiments and wrote the paper. GN participated in in vitro studies. S Amadio provided know-how and performed confocal fluorescence analysis. AS purified motor neurons and performed immunofluorescence studies. S Apolloni provided expertise and supervised in vivo experiments. PL provided expertise on motor neuron studies. CV designed, supervised the work and wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by N Bazan
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Parisi, C., Napoli, G., Amadio, S. et al. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ 23, 531–541 (2016). https://doi.org/10.1038/cdd.2015.153
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.153
This article is cited by
-
MicroRNA: Crucial modulator in purinergic signalling involved diseases
Purinergic Signalling (2023)
-
ISGylation is induced in neurons by demyelination driving ISG15-dependent microglial activation
Journal of Neuroinflammation (2022)
-
A Review of Molecular Interplay between Neurotrophins and miRNAs in Neuropsychological Disorders
Molecular Neurobiology (2022)
-
Research Progress on Exosomes and MicroRNAs in the Microenvironment of Postoperative Neurocognitive Disorders
Neurochemical Research (2022)
-
The role of microRNAs in diseases and related signaling pathways
Molecular Biology Reports (2022)
