
Abstract
Loss or silencing of tumor suppressors (TSs) promotes neoplastic transformation and malignant progression. To date, most work on TS has focused on their cell autonomous effects. Recent evidence, however, demonstrates an important noncell autonomous role for TS in the control of tumor-associated inflammation. We review evidence from clinical data sets and mouse model studies demonstrating enhanced inflammation and altered tumor microenvironment (TME) upon TS inactivation. We discuss clinical correlations between tumor-associated inflammation and inactivation of TS, and their therapeutic implications. This review sets forth the concept that TS can also suppress tumor-associated inflammation, a concept that provides new insights into tumor–host interactions. We also propose that in some cases the loss of TS function in cancer can be overcome through inhibition of the resulting inflammatory response, regardless whether it is a direct or an indirect consequence of TS loss.
Similar content being viewed by others


Facts
-
p53 mutations were documented in over 50% of human cancers. Loss of normal p53 function is frequently associated with an increased susceptibility to inflammasome-driven cancers such as ulcerative colitis-associated colorectal cancer.
-
In mouse cancer models of solid cancer, p53 mutations can cause chronic inflammation and persistent tissue damage.
-
Loss of adenomatous polyposis coli (APC) in human and mouse colon cancer is associated with enhanced infiltration of early adenomas by microbial products that elicit a tumor-associated inflammatory response by an upregulation of interleukin (IL)-17 that drives malignant progression.
-
Studies using mouse models reveal that loss of TGFβ signaling in cancer epithelial cells results in increased infiltration of inflammatory cells into the tumor microenvironment (TME).
-
Metformin, a widely prescribed antidiabetic drug with anti-inflammatory properties, is found to decrease the incidence of several malignancies including breast and pancreatic cancers.
Open Questions
-
Does inflammation have a critical role in p53 gain-of-function?
-
Does loss of tumor suppressor (TS) function result in the direct induction of inflammatory cytokines and chemokines or does it cause this effect due to enhanced tissue damage?
-
Would inhibition of tumor-associated inflammation provide an effective mean to overcome the loss of TS function?
TS are powerful transcriptional and signaling regulators that negatively modulate cell proliferation and survival. As such, TS counteract the growth promoting activity of oncogenes mainly through cell autonomous mechanisms. As amply described elsewhere,1 the major properties of classic TS genes are: (1) they are recessive and undergo biallellic inactivation in tumors; (2) inheritance of a single mutant allele increases cancer susceptibility as reflected by the autosomally dominant pattern of familial cancer syndromes; (3) TS genes are frequently inactivated in sporadic cancers. In addition to cell cycle progression and cell survival, TS regulate the detection and repair of DNA damage, protein turnover, autophagy, and metabolism. This review examines and discusses effects of TS on the interaction between the malignant cells and their microenvironment, a little studied aspect of TS function. We present evidence from mouse models and clinical studies that some TS also control tumor-elicited inflammation and will discuss potential mechanisms underlying this function. We hope that this review will lead to new thinking regarding the nonautonomous function of TS as negative regulator of inflammation. Accordingly, we suggest that anti-inflammatory therapies may partially compensate for loss of TS function in cancer.
TS as Regulators of Tumor-Associated Inflammation
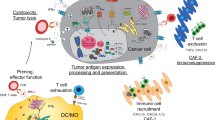
We postulate that one consequence of TS loss (Figure 1, input, black arrows), is elevated expression of growth factors, cytokines, and chemokines, which induce the recruitment, infiltration, and activation of host-derived inflammatory and stromal cells (Figure 1, mechanisms, in the big center circle). Once present within the tumor, these cells promote sustained cancer cell proliferation, evasion of apoptosis, replicative immortality, dysregulation of metabolism, invasion and metastasis, and genomic instability through a variety of noncell autonomous mechanisms (Figure 1, output, blue arrows). Collectively, these events create a pro-tumorigenic microenvironment that is immune suppressive and vascular permissive. Key studies supporting this hypothesis are listed in Table 1. It should be noted that it is not easy to distinguish the consequence of direct induction of inflammatory cytokines due to loss of TS function and induction of inflammatory cytokines during the tissue damage response that can also be triggered upon TS loss. It is perceivable that the two effects go hand in hand and that their actions are intertwined during tumor progression. For example, chronic inflammation associated with infection, autoimmune disease, prolonged exposure to environmental irritants or obesity precedes tumor development and can cause genomic instability, DNA damage, loss of TS function, early tumor promotion, and enhanced angiogenesis. The direct induction of inflammatory cytokines due to loss of TS function can further drive more tissue and cell damage associated with tumor initiation and progression.

TS regulate the inflammatory microenvironment: loss of TS (input, black arrows), including p53, TGFβ, APC, and PTEN, increases expression of growth factors, cytokines, and chemokines, which induce infiltration of host-derived inflammatory and stromal cells (mechanisms, in the big center circle). These cells in turn promote sustained proliferation, evasion of apoptosis, replicative immortality, dysregulation of metabolism, invasion, metastasis, and genomic instability (output, blue arrows)
p53
p53, the most extensively studied TS, provides a potent roadblock to tumorigenesis, and its loss has been documented in over 50% of human cancers. Mutant forms of p53 cannot cause cell cycle arrest and/or apoptotic cell death, no longer maintain genomic integrity, and fail to induce expression of metabolic regulators.2, 3 Clinical evidence suggests that p53 inactivation or deletion induces inflammation. For example, p53 is frequently mutated in ulcerative colitis-associated dysplastic lesions and colorectal cancer.4, 5 The increased frequency of p53 mutations in noncancerous ulcerative colitis epithelial cells may confer susceptibility to the development of colorectal cancer by enhancing the accumulation of nitric oxide (NO).6 In addition, a study of 833 patients with primary breast carcinoma revealed a strong association between p53 mutations and increased vascular endothelial growth factor (VEGF) expression.7 VEGF family members are expressed by tumor-infiltrating macrophages.8, 9 A study of head and neck squamous cell carcinoma (HNSCC) suggested that p53 suppresses inflammation through effects on NF-κB transcriptional activity.10
In mouse models, p53 mutations induce inflammation, bile duct injury, and fibrosis, which resemble the pathology of intrahepatic cholangiocarcinoma, a lethal human malignancy of the biliary epithelium, in which loss of p53 function is frequently observed.11 Mutant p53 forms (p53G515A or p53R172H) prolong TNF-induced NF-κB activation in cultured cells.12 As NF-κB contributes to elevated expression of inducible NO synthase (iNOS),13 this may explain the link between p53 mutations and NO accumulation. Even germ line p53 mutations can lead to chronic colonic inflammation and persistent tissue damage, thereby mimicking features of human colitis-associated colorectal cancer.12 In addition, p53 deletion induced an inflammatory microenvironment with elevated NO that accelerates spontaneous lymphoma development.14 In a mouse prostate cancer (PCa) model, loss of p53 results in enhanced transcription of cytokine and chemokine genes, accumulation of reactive oxygen species (ROS) and protein oxidation products, enhanced macrophage activation and neutrophil clearance, as well as high expression of inflammation markers.15 Combined loss of p53 and ATR, an essential factor in genome maintenance and DNA repair, leads to severe defects in hair follicle regeneration and deterioration of the intestinal epithelium, associated with tumor infiltration with inflammatory Mac1+Gr1+ cells.16 Specific p53 ablation in the intestinal epithelium resulted in increased permeability, leading to formation of an NF-κB-dependent inflammatory microenvironment and epithelial–mesenchymal transition.17 This effect is independent of the well-established role of p53 in cell cycle regulation, apoptosis, and senescence. More recently, ablation of p53 in hepatic stellate cells (HSCs) was found to increase liver fibrosis, and accelerate transformation of adjacent hepatocytes in a noncell autonomous manner involving macrophage polarization.18 Although p53 mutations were never found to occur in HSC or other types of nontransformed cells, these findings suggest a noncell autonomous function of p53 in the TME that can be manifested upon downregulation of p53 expression on account of gene methylation or other mechanisms. Reactivation of endogenous p53 in p53-deficient tumors in a mouse model of hepatocellular carcinoma (HCC) can cause complete tumor regressions.19 The underlying mechanism does not seem to depend on apoptosis, and instead may involve the induction of a cellular senescence program that is associated with upregulation of certain inflammatory cytokines.19
Mutant forms of p53 exhibit different modes of action.20 Whereas most mutants exhibit loss-of-TS functions, certain p53 mutants have gained oncogenic activity.21 Often, mutant p53 can antagonize the TS function of WT p53 in a dominant negative manner. Such a situation, however, is often transient, and is frequently followed by loss of heterozygosity (LOH) during cancer progression.20 Interestingly, gain-of-function p53 mutants, such as p53R273H, was found capable of NF-κB activation and can therefore induce tissue damage and chronic inflammation that promotes tumor development.12 These studies suggest that ability to induce inflammation is an important property that contributes to the oncogenic function of such p53 mutants. Two other members of the p53 family, p63 and p73, are also involved in the regulation of inflammation and cancer development.22, 23, 24 In summary, we suggest that the ability of WT p53 to inhibit tumor-associated inflammation should be added to the list of its tumor-suppressive activities.25
APC (adenomatous polyposis coli)
The APC tumor suppressor is particularly important in regulation of gut epithelial cell proliferation, adhesion, and migration. Germ line allelic loss of APC is a hallmark of human familial adenomatous polyposis syndrome as well as sporadic colorectal cancer. Studies suggest a strong correlation between colon cancer progression and tumor-associated inflammation.26, 27 Colorectal tumors exhibit immune/inflammatory infiltrates and inflammatory gene signature. In fact the type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Although CD4+ TH1 cells and CD8+ cytotoxic T cells (CTL) constitute a positive prognostic sign in human colorectal cancer,28 myeloid cells and T-helper IL-17-producing cells promote tumor progression.27 A clinical study demonstrated that upregulation of IL-17 negatively correlates with patient survival.29 As APC is a hallmark of colon cancer, these clinical studies suggest a likely association between APC deficiency and inflammation. Indeed, mouse model studies strongly support this link. For example, mono-allelic deletion of Apc in mouse intestinal epithelial cells (Apc+/Δ) results in tumor development upon inactivation of the WT Apc allele due to LOH.30, 31, 32 When Apc+/Δ mice are crossed with transgenic mice expressing constitutively active IκB kinase β (IKKβ) in intestinal epithelial cells (IECs), the compound mice exhibit more β-catenin positive (+) early lesions and small intestinal and colonic tumors relative to the parental Apc+/Δ line, and their survival is compromised.13 Accelerated tumorigenesis in this model is due to elevated expression of iNOS, leading to the formation of oxidative DNA lesions. Treatment of compound mice with an iNOS inhibitor decreased DNA damage markers and reduced early β-catenin (+) lesions and tumor load.13 Importantly, APC deficiency also induces upregulation of inflammatory cytokines. In a mouse model (CPC-APC) of colorectal tumorigenesis, in which Apc allelic loss is driven by Cdx2-Cre transgene, the resulting colorectal tumors exhibited upregulation of several proinflammatory cytokines, including IL-23 and IL-17A.33 IL-23 is expressed in tumor-infiltrating myeloid cells in response to invasion of the APC-deficient adenomas by microbial products.33 In turn, IL-23 induces the production of IL-17 by T cells and innate lymphoid cells leading to accelerated tumor growth and progression upon activation of IL-17 receptor A. Loss of Apc in this model (which is also due to LOH) results in rapid epithelial barrier deterioration, thus providing a major impetus for tumor-elicited inflammation.33 Furthermore, epithelial barrier disruption and signs of microbial invasion were also detected in human colorectal tumors,33 in which elevated IL-17 expression is a particularly bad prognostic sign.29 Another important inflammatory mediator is cyclooxygenase 2 (COX-2), which is critical for production of prostaglandin E2 that promotes neoplastic transformation of gut epithelial cells. Non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin show strong preventive effect and were recently proposed as an adjuvant therapy for colorectal cancer.34
PTEN (phosphatase and tensin homolog)
PTEN is a major TS that possesses dual lipid and protein phosphatase activity, and negatively regulates the pro-tumorigenic PI3K–AKT pathway.35, 36 A correlation between PTEN inactivation and tumor inflammation has not been carefully investigated. However, human HNSCC, are often deficient in PTEN, and show infiltration of inflammatory monocytes, angiogenesis and NF-κB activation, which were recapitulated in mice deficient in Pten and Tgfbr1.37 It should be noted, however, that loss of Tgfbr1 can also contribute to the inflammatory response seen in these mice. In addition, PTEN deficiency was reported to promote activation of the NF-κB network in a mouse model of pancreatic ductal adenocarcinoma driven by oncogenic KRAS, with accompanying stromal activation and immune cell infiltration.38 In addition to PTEN deficiency in epithelial cells, a number of recent studies suggest an important role of PTEN in stromal fibroblasts in regulation of the TME in breast cancer,39 further discussed below.
Collectively, such findings suggest that classical TS also function as negative regulators of inflammation. Their deletion, mutation or downregulation promotes the development of an inflammatory TME. Chronic and localized inflammation is one of the hallmarks of the cancer,40 and is a critical participant in tumor initiation and progression. Different from acute inflammation that can clear infection, heal wounds, and maintain tissue homeostasis, tumor-associated inflammation is often low in grade and chronic. Tumor-infiltrating inflammatory cells, together with an abundance of cancer-associated fibroblasts (CAFs) and endothelial progenitor cells (EPCs) create a microenvironment that favors tumor progression by providing growth factors, pro-angiogenic factors, proteases, and adhesion molecules.26, 41, 42 The inflammatory microenvironment also provides a selective pressure for emergence of malignant cell variants by enhancing genomic instability and heterogeneity, and epigenetic alterations.43
Downregulation of TS Activities in Stromal Fibroblasts
CAFs have important tumorigenic functions.44 In invasive human breast carcinomas, tumor-infiltrating CAFs produce SDF-1/CXCL12, which signals through its cognate receptor, CXCR4 on malignant cells to promote tumor growth, invasion, and angiogenesis.45 Gene expression signatures of the tumor stroma can predict clinical outcome in breast cancer.46 Epithelial and stromal cell signaling influence one another, and may co-evolve during the course of tumor progression.39 The dependence of tumors on stromal cells is also well echoed in lymphoma and multiple myeloma. Despite accumulation of numerous genetic alterations, the long-term culture of early-stage tumor cells without a stromal support is rarely successful. In a mouse model of B-cell lymphoma, Hedgehog ligands are secreted by stromal cells and function as survival factors for malignant progression through downregulation of Bcl2 in tumor cells.47 Another such factor is B-cell activating factor (BAFF), which is produced by nurse-like cells, specialized stromal cells that reside in the bone marrow. BAFF enhances chronic lymphocytic leukemia cell expression of c-MYC by activating canonical IKK/NF-κB signaling.48
Although mutations in TS genes were reported to occur within nonmalignant TME cells, including CAFs, such findings remain highly controversial.49 A few clinical studies reported p53 mutations in the stromal compartment of breast cancer,50, 51 but subsequent studies failed to reproduce these results.52, 53 Nevertheless, stromal-specific downregulation of TS gene expression or function could contribute to the malignant progression of epithelial tumors. Experimental approaches using mouse models in which TS genes have been inactivated in stromal cells show what can happen when TS are downregulated in nontransformed cells. Curiously, loss of p53 in stromal fibroblasts was found to result in loss of p53 in PCa cells.54 Loss of PTEN in stromal fibroblasts, leads to an increased PCa cell proliferation and inflammatory cell infiltration.55 In a mammary cancer model, genetic inactivation of PTEN in stromal cells increased expression, phosphorylation and recruitment of Ets2, and accelerated the transformation of mammary epithelial cells, resulting in tumors with extracellular matrix (ECM) remodeling, inflammatory infiltrates, and increased angiogenic capacity.56 A PTEN-specific signature was also observed in laser-captured human breast cancer stroma.56 In addition, PTEN loss decreased expression of miR-320, a micro RNA that targets ETS2 mRNA. This resulted in induction of an oncogenic secretome (for example, matrix metalloproteinase-9, bone morphogenetic protein-1, thrombospondin-1, and cathepsin B), that promotes tumor angiogenesis and invasion.57 Deletion of TGFβ type II receptor (TGFβRII) in FSP1+ stromal cells (Tgfbr2fspKO) contributes to epithelial transformation and results in development of invasive squamous cell carcinoma in the mouse forestomach.58, 59 The underlying mechanisms include hepatocyte growth factor,59 and altered p15, 16, and p21 expression mediated by inflammation-induced epigenetic regulation.58, 59 All of these studies support a general function of TS in regulation of the TME. However, it should be noted that the effect of stromal cells on the epithelial compartment of tumors is context dependent. For example, loss of Rb1 and Trp53 in the lung epithelium activates the epithelial cell-intrinsic Hedgehog signaling pathway and induces small-cell lung cancer, independent of the lung microenvironment.60 Different results were also obtained in which TGFβ signaling in CAF was found to be essential for induction of chemokines that recruit tumor-promoting B lymphocytes.61 The basis for these marked discrepancies is currently unknown.
Anti-Inflammatory Mechanisms Involving TS
As illustrated above, deletion of TS genes in epithelial cells or stromal fibroblasts can lead to induction of proinflammatory and pro-tumorigenic gene transcription. Several transcription factors are likely to be involved in this response, including NF-κB, a key regulator of inflammation.26, 62, 63 However, the pro-tumorigenic function of NF-κB is quite complex. Inactivation of IKKβ, a protein kinase needed for NF-κB activation, in IEC, does not result in direct anti-inflammatory effect although it prevents colitis-associated cancer by rendering IKKβ-deficient IECs susceptible to tumor-suppressive apoptosis. Specific deletion of IKKβ in myeloid cells, however, results in a significant decrease in tumor growth by preventing the production of proinflammatory cytokines that serve as growth factors. Thus, specific inactivation of IKKβ-dependent NF-κB signaling in two different cell types can attenuate inflammation-driven tumorigenesis.64 In a mouse model of lung adenocarcinoma, concomitant loss of p53 and expression of oncogenic KRASG12D resulted in NF-κB activation, whereas restoration of p53 expression led to NF-κB inhibition.65 Importantly, inhibition of NF-κB signaling induced apoptosis in p53-null lung cancer cell lines and inhibited their tumorigenic growth.65 NF-κB is a critical target for the inflammation suppressive activity of p53.66 WT p53 and NF-κB antagonize each other’s activities,66 and thus it is not surprising that p53 loss augments NF-κB activity. A recent study showed that glucocorticoid-induced repression of NF-κB targeted gene transcription was impaired upon p53 inactivation.67 LZAP, a putative TS, lost in ∼30% of human HNSCC, can also inhibit NF-κB, independently of ARF or p53.68 Components of the NF-κB system can also interact with other TS. IKKα activation in prostatic epithelial tumor cells, in response to lymphotoxin (LT) or RANK ligand produced by tumor-infiltrating immune cells, inhibits transcription of the metastasis suppressor Maspin, thereby promoting metastatic progression.69, 70 The cylindromatosis protein (CYLD), functions as deubiquitinase that removes lysine 63-linked ubiquitin chains from TRAF2, thereby inhibiting NF-κB activation.71 The gene CYLD is mutated in familial cylindromatosis, an autosomal dominant disease characterized by benign tumors derived from cells of skin appendages72 and thus it can be considered as a TS, CYLD inhibits tumor cell proliferation by blocking BCL-3-dependent NF-κB signaling.73 In the liver, however, disruption of the Cyld gene affected tumor progression through the protein kinases TAK1 and JNK.74
STAT3 is another important transcriptional regulator of cancer-associated inflammation. STAT3 dimers, induced upon phosphorylation by members of the JAK tyrosine kinase family, enter the nucleus and activate a broad array of target genes. STAT3 is activated in many cancers, including breast, pancreatic, colon and liver, and its conditional ablation inhibits cancer development and progression.75, 76, 77, 78, 79 Usually, STAT3 is activated by a number of cytokines, including IL-6. However, a number of nonconventional TS can negatively regulate STAT3, and their inactivation augments STAT3-mediated tumor-associated inflammation. For instance, deletion of Ptpn11, a gene encoding the tyrosine phosphatases Shp2, was found to promote STAT3 signaling resulting in liver hyperplasia and tumor development.75 This result suggests a TS function for Shp2 in hepatocellular carcinogenesis, yet Ptpn11 mutations remain to be identified, although Shp2 enzymatic activity may be inhibited by ROS that accumulate during tumor development.80 Another negative regulator of STAT3 is SOCS3, a member of the suppressor of cytokine signaling (SOCS) family that is often downregulated in human cancers.81 Conditional deletion of Socs3 gene in hepatocytes results in prolonged activation of STAT3 and accelerated HCC development.82, 83, 84
Inflammation and the Switch in TGFβ Function from a TS to a Metastasis Promoter
Transforming growth factor β (TGFβ) is both a TS and a tumor promoter, but the mechanisms underlying this switch in function are poorly understood. Recent studies suggest that inflammation may have an instrumental role in this process.85, 86 TGFβ has potent anti-inflammatory activity; loss or downregulation of TGFβ signaling induces inflammatory cell infiltration.85, 86 Specific deletion of Tgfbr2 in different epithelial cells including mammary, pancreatic, intestinal, colon, and head and neck squamous cell carcinomas, accelerates malignant progression and metastasis through effects on inflammation and the TME.86 Deletion of SMAD4 in colon epithelial cells results in an increased recruitment of CCR1+ myeloid cells (CD34+) that promote tumor invasion.87 Inactivation of TGFβ signaling in CAFs also results in expression of proinflammatory genes whose products promote tumor development.58, 59 Deletion of SMAD4 in T lymphocytes increases expression of IL-5, IL-6, and IL-13, and results in tumor infiltration with inflammatory cells, thereby promoting development of gastrointestinal carcinomas.88 However, it should be noted that in PCa, inactivation of TGFβ signaling in T cells resulted in tumor rejection.89 Curiously, epithelial-specific deletion of Smad4 did not result in the aforementioned forestomach tumor phenotype.88 Furthermore, TGFβ signaling is needed for CAF activation in PCa and production of chemokines, such as CXCL13, that recruit pro-tumoral B lymphocytes (Ammirante et al, manuscript in preparation) (Figure 2). Indeed, CXCL13 expression was found to correlate with clinical grade in human PCa.90 Moreover, TGFβ may have an important role in the pre-metastatic lung through S100A8 and S100A9, and Mac1+ myeloid cell recruitment.91 These data underscore the context-dependent nature of TGFβ signaling in cancer and its complexity.

Different mechanisms involving TME components contribute to T-cell tolerance and drug resistance in PCa. Androgen ablation in the TRAM mouse model induces cancer cell death/hypoxia and myofibroblast transdifferentiation (CAF). CAFs produce high levels of TGFβ and suppress host immunity by promoting Treg function and suppressing CTL activation. In addition, CAFs produce chemokines such as CXCL13, and recruit LT-producing B cells, a process dependent on TGFβ signaling in CAFs. Pro-tumorigenic B cells produce LT and immune-suppressive factors that have an important role in resistance of androgen ablation and chemotherapy
Some of the effects of TGFβ on inflammation and the TME may be exerted through NF-κB. For instance, TGFβ1 negatively regulates NF-κB activation through SMAD7,92 thus blocking proinflammatory TNF signals.93 Mice deficient in SMAD3 and infected with Helicobacter develop colon cancer due to increased inflammation.94 In TGFβ1-deficient mice, inflammation causes precancerous lesions to progress into colon cancer.95 In addition, TGFβ cross-talks with inflammatory pathways through modulation of IL-1 signaling.96 However, contradictory to these observations, over-expression of TGFβ1 in HNSCC epithelial cells results in inflammation, angiogenesis, and hyperproliferation.97 It is unclear what underlies these different observations, or the mechanisms that account for the proinflammatory effects of TGFβ. It is possible that TGFβ simply exerts its proinflammatory effects through chemokine production.98
Some of our work reveals that TGFβ regulates production of chemokine/chemokine receptors that are important for inflammatory cell recruitment. This includes stromal-derived factor 1 (SDF-1 or CXCL12), which mediates its effects through CXCR4, a receptor that is highly expressed on putative stem and progenitor cells.41, 99 TGFβ also suppresses CXCL1 and CXCL5, and inhibition of TGFβ signaling in cancer cells significantly increases expression of both chemokines.85 These chemokines are responsible for the recruitment of Gr-1+CD11b+ myeloid cells to the TME, where they produce large quantities of matrix metalloproteases. Several MMPs including MMP2 and MMP9 are important in the proteolytic activation of TGFβ. Interestingly, tumors with inactivated TGFβ signaling produce significantly higher levels of TGFβ1 than control tumors, probably due to increased infiltration of Gr-1+CD11b+ myeloid cells (Figure 3). Importantly, myeloid-specific inactivation of TGFβ signaling inhibited cancer metastasis.100 These data suggest that TGFβ signaling in cancer epithelial cells functions as TS through inhibition of inflammation, whereas myeloid TGFβ signaling in immature myeloid cell is metastasis promoting.

Tumor-associated inflammation is critical for converting TGFβ from a TS to a metastasis promoter. Host-derived immature myeloid Gr-1+CD11b+ cells are recruited into the TME upon deletion of Tgfbr2 in mammary carcinomas epithelial cells, through CXCL5:CXCR2 and SDF-1:CXCR4 interactions. In addition, Gr-1+CD11b+ cells express MMPs and TGFβ1, which promote tumor invasion and immune suppression. The effect of these immature myeloid cells on the TME and host immune surveillance promotes metastatic spread and is an essential contributor to the pro-metastatic action of TGFβ
Clinical Correlations and Therapeutic Implications
The targeting of the inflammatory TME may be an effective therapeutic option for patients whose cancers display inactivation or decreased expression of TS genes that have anti-inflammatory activity. As discussed above, inactivation or downregulation of certain TS genes can result in increased infiltration of inflammatory/immune cells and CAFs into the TME. As this process can make important contributions to cancer development and progression, we suggest that it represents an attractive target for therapeutic intervention. With the exception of compounds that target the Mdm2:p53 interaction,101 it is difficult or even impossible to design small molecules that directly restore TS function. However, there is ample evidence for the association between TS loss and enhanced tumoral inflammation, which can be targeted by various auto-inflammatory drugs. Several targeting opportunities are envisioned (Figure 4), including IKKβ or IKKα inhibitors, metformin, as well as NSAIDs. In addition, neutralizing antibodies and antagonists may offer additional options to inhibit the inflammatory TME and enhance antitumor immunity. Furthermore, cell type-specific targeting may also provide alternative, including tumor-associated macrophages,102, 103 Gr-1+CD11b+ immature myeloid cells or myeloid-derived suppressor cells,100, 104 and B cells.61 This is demonstrated in case of TGFβ, in which myeloid-specific deletion of TGFβ signaling diminished cancer metastasis in a number of mouse models (Figure 2).100

Targeting opportunities aimed at inflammation and the TME in case of TS loss. IKKβ or IKKα inhibitors, metformin, as well as anti-inflammation drugs can be used with conventional chemotherapeutic agents, radiation therapies, and targeted therapies. In addition, neutralization antibodies, antagonists, and cell type-specific targeting may offer additional options to inhibit the inflammatory TME and enhance host antitumor immunity
Clinical studies support strategies aimed at inflammation and its consequences. For example, a study of 833 patients with primary breast carcinoma revealed a strong association between p53 mutations and increased VEGF expression,7 thus such patients may respond to inhibitors of VEGF synthesis and signaling. As VEGF family members are expressed by tumor-infiltrating macrophages,8 targeting inflammation may provide additional therapeutic options. Another retrospective study of HNSCC suggested that p53 suppresses inflammation through its effect on NF-κB activity and gene expression.105 Stable cell lines expressing the R175H, R273H, or D281G mutant forms of p53 exhibit loss of drug sensitivity due to activation of alternative NF-κB2 signaling.106 Targeting the connection between p53 and NF-κB may increase chemo-sensitivity and reduce associated inflammation, and it may be done through the use of IKKβ or IKKα inhibitors. Drugs that inhibit inflammation may restore the anti-inflammatory properties of TS and facilitate tumor regression by enhancing antitumor immunity. Several existing drugs may be considered as candidates for such an approach, along with more classical or targeted anticancer therapies. Metformin, a widely prescribed antidiabetic drug, has documented anti-inflammatory properties. When used in postmenopausal women with type II diabetes, metformin may reduce the incidence of invasive breast cancer.107 Aspirin demonstrates strong preventive effects in many different cancers.108 CXCL13 antagonists or B-cell-depleting drugs may prevent the development of castrate resistant PCa.61 Unlike conventional chemotherapeutic agents, radiation therapy, and targeted therapies, the inhibition of tumoral inflammation may avoid the all too frequent development of drug resistance. When combined with classic targeting strategies aimed at genetic and epigenetic alterations in cancer cells, therapies aimed at the inflammatory TME and enhancement of antitumor immunity should offer more specific and efficient outcomes that, in addition to cancer cells, also target extrinsic processes that depend on cells with stable genomes, devoid of gate keeper mutations. However, a major challenge is to differentiate host-protective acute inflammation from tumor-promoting chronic inflammation as well as the opposing and hard to predict outcomes of signaling network modulation, such as TGFβ systems in different cells and tumor types.
Conclusions and Perspectives
Suppression of inflammatory cytokine/chemokine production is a noncell autonomous process that contributes to overall tumor suppression. Loss of TS function results in an inflammatory response that is supposed to be beneficial for cell/tissue damage repair. However, loss of TS function can also have deleterious effects on the tissue and tumor microenvironment. The synergy between an altered microenvironment and the genetic alterations that are directly acquired by cancer cells allows these cells to evade various tumor surveillance mechanisms and become fully malignant. How can the inflammatory response be both good and bad? The idea that processes can have such dual effects is consistent with a major evolutionary theory termed antagonistic pleiotropy by Williams GC.
TSs confer protection against cancer by blocking the abnormal proliferation of cells. Loss of TS function results in uncontrolled epithelial cell division and induction of tumor-associated inflammation. The behavior of tumor-initiating cells that have just lost TS function is affected by the dynamic microenvironment in which they live, which is full of growth factors, chemokines, inflammatory cytokines, and reactive oxygen/nitrogen species. This environment also contains inflammatory cells that produce factors that stimulate cancer growth as well as immune cells that can eliminate malignant cells. How TS nonautonomous molecular signature is activated and maintained, and how it influences malignant progression, as well as immune evasion are all open questions that should be addressed in future studies. Nonetheless, even with our current limited knowledge, potential new strategies for cancer therapies can be envisioned. For example, various auto-inflammatory drugs, IKKβ or IKKα inhibitors, metformin, as well as NSAIDs may help restore TS function.
Abbreviations
- TS:
-
tumor suppressors
- TME:
-
tumor microenvironment
- NO:
-
nitric oxide
- VEGF:
-
vascular endothelial growth factor
- HNSCC:
-
head and neck squamous cell carcinomas
- iNOS:
-
nitric oxide synthase
- PCa:
-
prostate cancer
- ROS:
-
reactive oxygen species
- HSC:
-
hepatic stellate cells
- APC:
-
adenomatous polyposis coli
- CTL:
-
cytotoxic T cells
- LOH:
-
loss of heterozygocity
- IECs:
-
intestinal epithelial cells
- COX-2:
-
cyclooxygenase 2
- NSAIDS:
-
non-steroid anti-inflammation drugs
- PTEN:
-
phosphatase and tensin homolog
- CAFs:
-
cancer-associated fibroblasts
- EPCs:
-
endothelial progenitor cells
- BAFF:
-
B-cell activating factor
- ECM:
-
extracellular matrix
- TGFβRII:
-
TGFβ type II receptor
- FSP1+:
-
fibroblast specific protein 1
- Tgfbr2fspKO:
-
mice with Tgfbr2 deletion in stromal cells expressing fibroblast specific protein 1
- LT:
-
lymphotoxin
- CYLD:
-
cylindromatosis protein
- SOCS:
-
suppressor of cytokine signaling
- TGFβ:
-
transforming growth factor β
- HCC:
-
hepatocellular carcinoma
References
Sherr CJ . Principles of tumor suppression. Cell 2004; 116: 235–246.
Hainaut P, Hollstein M . p53 and human cancer: the first ten thousand mutations. Adv Cancer Res 2000; 77: 81–137.
Kruse JP, Gu W . Modes of p53 regulation. Cell 2009; 137: 609–622.
Brentnall TA, Crispin DA, Rabinovitch PS, Haggitt RC, Rubin CE, Stevens AC et al. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology 1994; 107: 369–378.
Yin J, Harpaz N, Tong Y, Huang Y, Laurin J, Greenwald BD et al. p53 point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 1993; 104: 1633–1639.
Hussain SP, Amstad P, Raja K, Ambs S, Nagashima M, Bennett WP et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res 2000; 60: 3333–3337.
Linderholm B, Lindh B, Tavelin B, Grankvist K, Henriksson R . p53 and vascular-endothelial-growth-factor (VEGF) expression predicts outcome in 833 patients with primary breast carcinoma. Int J Cancer 2000; 89: 51–62.
Monnier Y, Zaric J, Ruegg C . Inhibition of angiogenesis by non-steroidal anti-inflammatory drugs: from the bench to the bedside and back. Curr Drug Targets Inflamm Allergy 2005; 4: 31–38.
Salvado MD, Alfranca A, Haeggstrom JZ, Redondo JM . Prostanoids in tumor angiogenesis: therapeutic intervention beyond COX-2. TrendsMol Med 2012; 18: 233–243.
Lee TL, Yang XP, Yan B, Friedman J, Duggal P, Bagain L et al. A novel nuclear factor-kappaB gene signature is differentially expressed in head and neck squamous cell carcinomas in association with TP53 status. Clin Cancer Res 2007; 13: 5680–5691.
Farazi PA, Zeisberg M, Glickman J, Zhang Y, Kalluri R, DePinho RA . Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res 2006; 66: 6622–6627.
Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S et al. Mutant p53 prolongs nf-kappab activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013; 23: 634–646.
Shaked H, Hofseth LJ, Chumanevich A, Chumanevich AA, Wang J, Wang Y et al. Chronic epithelial NF-kappaB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc Natl Acad Sci USA 2012; 109: 14007–14012.
Hussain SP, He P, Subleski J, Hofseth LJ, Trivers GE, Mechanic L et al. Nitric oxide is a key component in inflammation-accelerated tumorigenesis. Cancer Res 2008; 68: 7130–7136.
Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG et al. p53 is a suppressor of inflammatory response in mice. FASEB J 2005; 19: 1030–1032.
Ruzankina Y, Schoppy DW, Asare A, Clark CE, Vonderheide RH, Brown EJ . Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nat Genet 2009; 41: 1144–1149.
Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013; 23: 93–106.
Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE et al. Non-cell-autonomous tumor suppression by p53. Cell 2013; 153: 449–460.
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445: 656–660.
Vousden KH, Prives C . Blinded by the light: the growing complexity of p53. Cell 2009; 137: 413–431.
Brosh R, Rotter V . When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9: 701–713.
Lu H, Yang X, Duggal P, Allen CT, Yan B, Cohen J et al2011 TNF-alpha promotes c-REL/DeltaNp63alpha interaction and TAp73 dissociation from key genes that mediate growth arrest and apoptosis in head and neck cancer. Cancer Res 71: 6867–6877.
Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22: 2677–2691.
Yang X, Lu H, Yan B, Romano RA, Bian Y, Friedman J et al. DeltaNp63 versatilely regulates a broad NF-kappaB gene program and promotes squamous epithelial proliferation, migration, and inflammation. Cancer Res 2011; 71: 3688–3700.
Bieging KT, Mello SS, Attardi LD . Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014; 14: 359–370.
Grivennikov SI, Greten FR, Karin M . Immunity, inflammation, and cancer. Cell 2010; 140: 883–899.
Mantovani A, Allavena P, Sica A, Balkwill F . Cancer-related inflammation. Nature 2008; 454: 436–444.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–1964.
Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 2011; 71: 1263–1271.
Amos-Landgraf JM, Irving AA, Hartman C, Hunter A, Laube B, Chen X et al. Monoallelic silencing and haploinsufficiency in early murine intestinal neoplasms. Proc Natl Acad Sci USA 2012; 109: 2060–2065.
Bilger A, Shoemaker AR, Gould KA, Dove WF . Manipulation of the mouse germline in the study of Min-induced neoplasia. Semin Cancer Biol 1996; 7: 249–260.
Merritt AJ, Gould KA, Dove WF . Polyclonal structure of intestinal adenomas in ApcMin/+ mice with concomitant loss of Apc+ from all tumor lineages. Proc Natl Acad Sci USA 1997; 94: 13927–13931.
Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012; 491: 254–258.
Chia WK, Ali R, Toh HC . Aspirin as adjuvant therapy for colorectal cancer—reinterpreting paradigms. Nat Rev Clin Oncol 2012; 9: 561–570.
Song MS, Salmena L, Pandolfi PP . The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012; 13: 283–296.
Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998; 95: 29–39.
Bian Y, Hall B, Sun ZJ, Molinolo A, Chen W, Gutkind JS et al. Loss of TGF-beta signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012; 31: 3322–3332.
Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer discovery 2011; 1: 158–169.
Wallace JA, Li F, Leone G, Ostrowski MC . Pten in the breast tumor microenvironment: modeling tumor-stroma coevolution. Cancer Res 2011; 71: 1203–1207.
Hanahan D, W RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Balkwill F, Coussens LM . Cancer: an inflammatory link. Nature 2004; 431: 405–406.
Coussens LM, Werb Z . Inflammation and cancer. Nature 2002; 420: 860–867.
Bristow RG, Hill RP . Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer 2008; 8: 180–192.
Kalluri R, Zeisberg M . Fibroblasts in cancer. Nat Rev Cancer 2006; 6: 392–401.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005; 121: 335–348.
Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 2008; 14: 518–527.
Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med 2007; 13: 944–951.
Zhang W, Kater AP, Widhopf GF 2nd, Chuang HY, Enzler T, James DF et al. B-cell activating factor and v-Myc myelocytomatosis viral oncogene homolog (c-Myc) influence progression of chronic lymphocytic leukemia. Proc Natl Acad Sci USA 2010; 107: 18956–18960.
Weinberg RA . Coevolution in the tumor microenvironment. Nat Genet 2008; 40: 494–495.
Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C . Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet 2002; 32: 355–357.
Patocs A, Zhang L, Xu Y, Weber F, Caldes T, Mutter GL et al. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med 2007; 357: 2543–2551.
Campbell IG, Qiu W, Polyak K, Haviv I . Breast-cancer stromal cells with TP53 mutations. N Engl J Med 2008 author reply 358: 1634–1635.
Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet 2008; 40: 650–655.
Hill R, Song Y, Cardiff RD, Van Dyke T . Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 2005; 123: 1001–1011.
Svensson RU, Haverkamp JM, Thedens DR, Cohen MB, Ratliff TL, Henry MD . Slow disease progression in a C57BL/6 pten-deficient mouse model of prostate cancer. Am J Pathol 2011; 179: 502–512.
Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009; 461: 1084–1091.
Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol 2012; 14: 159–167.
Achyut BR, Bader DA, Robles AI, Wangsa D, Harris CC, Ried T et al. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-beta signaling. PLoS Genet 2013; 9: e1003251.
Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004; 303: 848–851.
Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med 2011; 17: 1504–1508.
Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M . B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010; 464: 302–305.
Ben-Neriah Y, Karin M . Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol 2011; 12: 715–723.
Karin M, Greten FR . NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005; 5: 749–759.
Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004; 118: 285–296.
Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009; 462: 104–107.
Perkins ND . Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 2007; 8: 49–62.
Murphy SH, Suzuki K, Downes M, Welch GL, De Jesus P, Miraglia LJ et al. Tumor suppressor protein (p)53, is a regulator of NF-kappaB repression by the glucocorticoid receptor. Proc Natl Acad Sci USA 2011; 108: 17117–17122.
Wang J, An H, Mayo MW, Baldwin AS, Yarbrough WG . LZAP, a putative tumor suppressor, selectively inhibits NF-kappaB. Cancer Cell 2007; 12: 239–251.
Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL et al. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 2007; 446: 690–694.
Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011; 470: 548–553.
Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G . The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003; 424: 801–805.
Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 2000; 25: 160–165.
Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R . Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell 2006; 125: 665–677.
Nikolaou K, Tsagaratou A, Eftychi C, Kollias G, Mosialos G, Talianidis I . Inactivation of the deubiquitinase CYLD in hepatocytes causes apoptosis, inflammation, fibrosis, and cancer. Cancer Cell 2012; 21: 738–750.
Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 2011; 19: 629–639.
Fukuda A, Wang SC, JPt Morris, Folias AE, Liou A, Kim GE et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011; 19: 441–455.
Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G et al. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 2006; 126: 489–502.
Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011; 19: 456–469.
Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013; 23: 107–120.
He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010; 17: 286–297.
Li Y, de Haar C, Peppelenbosch MP, van der Woude CJ . SOCS3 in immune regulation of inflammatory bowel disease and inflammatory bowel disease-related cancer. Cytokine Growth Factor Rev 2012; 23: 127–138.
Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 2003; 4: 540–545.
Ogata H, Chinen T, Yoshida T, Kinjyo I, Takaesu G, Shiraishi H et al. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene 2006; 25: 2520–2530.
Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, Mueller SC et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci USA 2008; 105: 2445–2450.
Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M et al. Abrogation of TGFbeta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008; 13: 23–35.
Yang L . TGFbeta and cancer metastasis: an inflammation link. Cancer Metastasis Rev 2010; 29: 263–271.
Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H et al. SMAD4-deficient intestinal tumors recruit CCR1(+) myeloid cells that promote invasion. Nat Genet 2007; 39: 467–475.
Kim BG, Li C, Qiao W, Mamura M, Kasprzak B, Anver M et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature 2006; 441: 1015–1019.
Donkor MK, Sarkar A, Savage PA, Franklin RA, Johnson LK, Jungbluth AA et al. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-beta1 cytokine. Immunity 2011; 35: 123–134.
Singh S, Singh R, Sharma PK, Singh UP, Rai SN, Chung LW et al. Serum CXCL13 positively correlates with prostatic disease, prostate-specific antigen and mediates prostate cancer cell invasion, integrin clustering and cell adhesion. Cancer Lett 2009; 283: 29–35.
Hiratsuka S, Watanabe A, Aburatani H, Maru Y . Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol 2006; 8: 1369–1375.
Monteleone G, Mann J, Monteleone I, Vavassori P, Bremner R, Fantini M et al. A failure of transforming growth factor-beta1 negative regulation maintains sustained NF-kappaB activation in gut inflammation. J Biol Chem 2004; 279: 3925–3932.
Hong S, Lee C, Kim SJ . Smad7 sensitizes tumor necrosis factor induced apoptosis through the inhibition of antiapoptotic gene expression by suppressing activation of the nuclear factor-kappaB pathway. Cancer Res 2007; 67: 9577–9583.
Maggio-Price L, Treuting P, Zeng W, Tsang M, Bielefeldt-Ohmann H, Iritani BM . Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res 2006; 66: 828–838.
Engle SJ, Ormsby I, Pawlowski S, Boivin GP, Croft J, Balish E et al. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res 2002; 62: 6362–6366.
Lu T, Tian L, Han Y, Vogelbaum M, Stark GR . Dose-dependent cross-talk between the transforming growth factor-beta and interleukin-1 signaling pathways. Proc Natl Acad Sci USA 2007; 104: 4365–4370.
Lu SL, Reh D, Li AG, Woods J, Corless CL, Kulesz-Martin M et al. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res 2004; 64: 4405–4410.
Bierie B, Chung CH, Parker JS, Stover DG, Cheng N, Chytil A et al. Abrogation of TGF-beta signaling enhances chemokine production and correlates with prognosis in human breast cancer. J Clin Invest 2009; 119: 1571–1582.
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008; 13: 206–220.
Pang Y, Gara SK, Achyut BR, Li Z, Yan HH, Day CP et al. Transforming growth factor beta signaling in myeloid cells is required for tumor metastasis. Cancer discovery 2013; 3: 936–951.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844–848.
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013; 19: 1264–1272.
Qian BZ, Pollard JW . Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141: 39–51.
Gabrilovich DI, Nagaraj S . Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9: 162–174.
Ferris RL, Grandis JR . NF-kappaB gene signatures and p53 mutations in head and neck squamous cell carcinoma. Clin Cancer Res 2007; 13: 5663–5664.
Scian MJ, Stagliano KE, Anderson MA, Hassan S, Bowman M, Miles MF et al. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol Cell Biol 2005; 25: 10097–10110.
Chlebowski RT, McTiernan A, Wactawski-Wende J, Manson JE, Aragaki AK, Rohan T et al. Diabetes, metformin, and breast cancer in postmenopausal women. J Clin Oncol 2012; 30: 2844–2852.
Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW . Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 2011; 377: 31–41.
Farazi PA, Zeisberg M, Glickman J, Zhang Y, Kalluri R, DePinho RA . Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res 2006; 66: 6622–6627.
Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013; 23: 93–106.
Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE et al. Non-cell-autonomous tumor suppression by p53. Cell 2013; 153: 449–460.
Hill R, Song Y, Cardiff RD, Van Dyke T . Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 2005; 123: 1001–1011.
Chien WM, Garrison K, Caufield E, Orthel J, Dill J, Fero ML . Differential gene expression of p27Kip1 and Rb knockout pituitary tumors associated with altered growth and angiogenesis. Cell Cycle 2007; 6: 750–757.
Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012; 491: 254–258.
Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov 2011; 1: 158–169.
Bian Y, Hall B, Sun ZJ, Molinolo A, Chen W, Gutkind JS et al. Loss of TGF-beta signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012; 31: 3322–3332.
Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009; 461: 1084–1091.
Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M et al. Abrogation of TGFbeta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008; 13: 23–35.
Yang L . TGFbeta and cancer metastasis: an inflammation link. Cancer Metastasis Rev 2010; 29: 263–271.
Achyut BR, Bader DA, Robles AI, Wangsa D, Harris CC, Ried T et al. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-beta signaling. PLoS Genet 2013; 9: e1003251.
Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H et al. SMAD4-deficient intestinal tumors recruit CCR1(+) myeloid cells that promote invasion. Nat Genet 2007; 39: 467–475.
Kim BG, Li C, Qiao W, Mamura M, Kasprzak B, Anver M et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature 2006; 441: 1015–1019.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by G Melino
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Yang, L., Karin, M. Roles of tumor suppressors in regulating tumor-associated inflammation. Cell Death Differ 21, 1677–1686 (2014). https://doi.org/10.1038/cdd.2014.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.131
This article is cited by
-
TGFβ biology in cancer progression and immunotherapy
Nature Reviews Clinical Oncology (2021)
-
Activation of PAR2 by tissue factor induces the release of the PTEN from MAGI proteins and regulates PTEN and Akt activities
Scientific Reports (2020)
-
Novel tumor suppressor SPRYD4 inhibits tumor progression in hepatocellular carcinoma by inducing apoptotic cell death
Cellular Oncology (2019)
-
A mouse model of the Δ133p53 isoform: roles in cancer progression and inflammation
Mammalian Genome (2018)
-
Molecular spectrum of secretome regulates the relative hepatogenic potential of mesenchymal stem cells from bone marrow and dental tissue
Scientific Reports (2017)
