
Abstract
Cellular FLICE-inhibitory protein (c-FLIP) proteins are known as potent inhibitors of death receptor-mediated apoptosis by interfering with caspase-8 activation at the death-inducing signaling complex (DISC). Among the three human isoforms, c-FLIPlong, c-FLIPshort and c-FLIPR, the latter isoform is poorly characterized. We report here the characterization of murine c-FLIPR and show that it is the only short c-FLIP isoform expressed in mice. By generating several mutants, we demonstrate that both death effector domains (DEDs) are required for DISC binding and the antiapoptotic function of c-FLIPR. Surprisingly, the C-terminal tail is important for both protein stability and DISC recruitment. Three-dimensional modeling of c-FLIPR revealed a substantial similarity of the overall structures and potential interaction motifs with the viral FLIP MC159. We found, however, that c-FLIPR uses different structural motifs for its DISC recruitment. Whereas MC159 interferes with interaction and self-oligomerization of the DISC component FADD by its extensive hydrophilic surface, a narrow hydrophobic patch of c-FLIPR on the surface of DED2 is crucial for DISC association. Thus, despite the presence of similar tandem DEDs, viral and cellular FLIPs inhibit apoptosis by remarkably divergent mechanisms.
Similar content being viewed by others


Main
Death receptors like CD95 (Fas/Apo-1) transduce death signals upon ligand binding and formation of a death-inducing signaling complex (DISC), which comprises the receptor, the adaptor protein Fas-associated death domain (FADD) and procaspase-8 (FLICE) or procaspase-10.1 This assembly brings procaspase-8/10 in close proximity to the receptor and allows them to be activated by dimerization.2, 3, 4 Activation of the initiator caspases can be counteracted by FLICE-inhibitory proteins (FLIPs).5, 6 Next to viral FLIP (v-FLIP) proteins,7 three cellular homologs (cellular FLICE-inhibitory proteins (c-FLIPs)) have been identified, namely c-FLIPlong, c-FLIPshort and c-FLIPR, which are generated by differential splicing.8, 9, 10 The C-terminus of c-FLIPlong contains a catalytically inactive caspase-like domain, whereas c-FLIPshort and c-FLIPR have only a truncated C-terminus.
As a characteristic feature, FLIP proteins contain tandem death effector domains (DEDs), which mediate their recruitment into the DISC.10, 11 The DED forms a bundle of six antiparallel α-helices similar to the death domain (DD) and the caspase recruitment domain (CARD), two other signaling motifs involved in homotypic protein interactions.12 Curiously, despite their importance in apoptosis, there are currently no reported structures for cellular FLIP proteins. Only the structure of the tandem DEDs of the v-FLIP MC159 from Molluscum contagiosum has been reported showing a rigidly associated dumbbell-shaped molecule, which is tightly packed by an extensive hydrophobic interface between its two DEDs.13, 14 Similar to FADD, each DED of MC159 contains two prominent surface features important for protein interactions that distinguish them from other death motifs, namely a hydrophobic patch and a charge triad, also known as RxDL motif.13, 14, 15, 16
Although the structure of MC159 provides a template for understanding the mechanism of FLIP inhibition, it is unknown which structural motifs mediate DISC recruitment of cellular FLIP proteins. In the present study, we characterize for the first time the short isoform of murine c-FLIP and show that c-FLIPR but not c-FLIPshort is the sole truncated isoform expressed in murine cells. We further demonstrate that both DEDs of c-FLIPR are crucial for DISC recruitment and inhibition of caspase-8 processing. Moreover, we found that the unique C-terminal tail of c-FLIPR not only determines protein stability, but also its antiapoptotic function. Interestingly, despite the structural similarity of MC159 and c-FLIPR, point mutations in the charge triads, which block apoptosis protection by MC159, did not abrogate apoptosis inhibition by c-FLIPR. Instead, we show that the hydrophobic patch in DED2 is critical for recruitment of c-FLIPR into the DISC. Thus, our results demonstrate that viral and cellular FLIPs prevent apoptosis by divergent mechanisms, suggesting a surprisingly functional diversity of the DED motif.
Results
c-FLIPR but not c-FLIPshort is expressed in mouse
Although two short splice variants, c-FLIPshort and c-FLIPR, have been described in human cells,8, 10, 17 little is known about their expression in other species. As human c-FLIPshort is expressed primarily in lymphoid tissues,18 we prepared RNA from mouse thymus and performed RT-PCR. A cDNA with an open reading frame of 648 bp was cloned, encoding a protein of 215 amino acids (aa). Surprisingly, sequence alignment with human c-FLIP revealed that it resembles more human c-FLIPR than c-FLIPshort (Figure 1a).

Mouse and human c-FLIP isoforms. (a) Sequence alignment of murine c-FLIPR with human c-FLIPR and c-FLIPshort. The sequence of the short c-FLIP isoform cloned from mouse thymus was aligned to human c-FLIPR (as published in Djerbi et al.17) and human c-FLIPshort (AAC51623) using the Geneious software (http://www.geneious.com). Sequences are shown in single-letter amino-acid code. (b) Intron–exon structure of the human (top) and mouse (bottom) gene locus. Exons containing the translational start and stop codons are labeled with ‘atg’, ‘taa’ and ‘tga’, respectively. Regions encoding for the first DED, the second DED and the caspase-like domains are indicated. The human-specific exon 7, which is contained in the c-FLIPshort mRNA, is marked in red, and the intronic sequences are marked in green
Comparison of the gene structures showed that the human c-FLIP gene comprises 14 exons spanning about 52 kb, whereas the mouse gene has 10 exons spanning 42 kb (Figure 1b). The mRNA of mouse c-FLIPlong contains all 10 exons, with the translational start in exon 2 and the stop codon in exon 10 (Figure 1b). Mouse c-FLIPR is generated when exon 5 is not spliced to exon 6, which encodes a part of the c-FLIPlong protein. Instead, the c-FLIPR mRNA includes an intronic sequence with an in-frame stop codon (Figure 1b). Similar splice events occur during generation of human c-FLIPlong and c-FLIPR. However, the human c-FLIP gene contains an additional exon 7 that can be alternatively spliced to c-FLIPshort (Figure 1b). As a homologous exon is missing in the mouse gene, c-FLIPR is the only short isoform expressed in murine cells.
Mouse c-FLIPR is rapidly degraded via the proteasome pathway
To characterize mouse c-FLIPR, we generated Flag-tagged versions of the full-length molecule and a C-terminal deletion mutant (c-FLIPR-ΔC, aa 1–186) comprising only the tandem DEDs (Figure 2a). These constructs were stably transfected into the mouse B-cell line A20 that is sensitive to CD95-mediated apoptosis. Analyzing various clones with different expression levels, we generally observed a higher expression of the truncated c-FLIPR-ΔC construct compared with the full-length protein, although the expression of other DISC components, including CD95, FADD and caspase-8, remained unchanged (Figure 2b). As human c-FLIPshort and c-FLIPR have a short half-life time,8, 19 we tested the stability of mouse c-FLIPR. To this end, we treated A20 cells expressing comparable amounts of either c-FLIPR or c-FLIPR-ΔC with the translation inhibitor cycloheximide (CHX). As shown in Figure 2c, expression of full-length c-FLIPR decreased rapidly upon addition of CHX, suggesting a fast degradation of the protein. In contrast, the levels of c-FLIPR-ΔC remained constant even after 8 h of CHX treatment (Figure 2c). Thus, the C-terminal amino acids seem to determine the stability of the mouse c-FLIPR protein.

Deletion of the C-terminal tail of c-FLIPR increases protein stability. (a) Schematic representation of the different c-FLIPR mutants. Full-length murine c-FLIPR and the ΔC-construct contain the two characteristic DEDs at amino-acid positions 11–77 and 96–174. c-FLIPR-ΔC lacks the C-terminal tail starting at position 200 aa. The constructs DED1 and DED2 encode only the first and second DED, respectively. (b) A20 transfectants stably expressing an empty pEF-Flag-control vector, full-length c-FLIPR or c-FLIPR-ΔC cloned into the pEF-Flag-vector were analyzed by western blotting for the expression of c-FLIPR, CD95, caspase-8 and FADD. Blots were probed with either anti-β-actin or anti-tubulin to control equal protein loading. The results show representative blots from several generated clones with different expression levels of the transgenes. (c) A20 cells stably transfected either with full-length c-FLIPR or with c-FLIPR-ΔC were treated for 0, 2, 4, 6 and 8 h with CHX (10 μg/ml). Subsequently, cells were lysed and analyzed by western blotting for c-FLIPR expression. β-Actin served as loading control. (d) 293T cells were transiently transfected with His–ubiquitin or with His–ubiquitin and the indicated c-FLIPR constructs. After 20 h, the cells were incubated for another 4 h with 10 μg/ml of the proteasome inhibitor MG132. After cell lysis, proteins bound to His-tagged ubiquitin were precipitated with Ni-NTA agarose. Precipitates were analyzed for the respective c-FLIPR-constructs by western blotting. The partial binding of the non-ubiquitinated c-FLIPR versions to the Ni2+-beads is presumably due to the highly negatively charged epitope tag sequences
We next tested whether degradation of c-FLIPR is mediated by the ubiquitin–proteasome pathway. HEK293T cells were transfected with His-tagged ubiquitin alone or together with c-FLIPR, c-FLIPR-ΔC or additional mutants. Immmunoblot analysis of the ubiquitinated proteins, enriched from cell lysates with Ni2+-beads, revealed that c-FLIPR was polyubiquitinated, showing a typical high-molecular-weight ubiquitin ladder (Figure 2d). In contrast, ubiquitination of the mutant c-FLIPR-ΔC was strongly reduced, suggesting that the C-terminal sequence is important for this process.
Poukkula et al.20 found two lysine residues (K192, K195), which mediate ubiquitin-dependent degradation of human c-FLIPshort. To investigate whether these lysines are also required for degradation of mouse c-FLIPR, we mutated the homolog residues to arginine (K196R/K200R). Surprisingly, the K196R/K200R-mutant was as efficiently ubiquitinated as the wild-type protein (Figure 2d). Therefore, the C-terminal tail does not contain the essential ubiquitination sites but might rather play an indirect role for c-FLIPR degradation. Consistent with this, c-FLIPR mutants encoding only the first (DED1) or the second DED (DED2) became also ubiquitinated (Figure 2d). Thus, these experiments suggest that mouse c-FLIPR has a short half-life time due to proteasome-mediated degradation, which is influenced by its unique C-terminus.
Deletion of the C-terminal tail of c-FLIPR diminishes its antiapoptotic function
To investigate the antiapoptotic function of c-FLIPR, we treated wild-type A20 cells and transfectants stably expressing c-FLIPR constructs with CD95 ligand (CD95L) for up to 8 h, and measured caspase activation at various time points. As detected by their processing, activation of caspase-3 and caspase-8 was readily detected upon stimulation with CD95L in wild-type and vector control cells (Figure 3a). In contrast, both caspases remained uncleaved in cells expressing c-FLIPR-ΔC or c-FLIPR (Figure 3a). Thus, mouse c-FLIPR blocks CD95-mediated caspase activation.

Deletion of the C-terminal tail diminishes the antiapoptotic potential of c-FLIPR. (a) Wild-type A20 cells (wt) or A20 cells stably transfected with either full-length c-FLIPR, c-FLIPR-ΔC or an empty vector control were stimulated for the indicated time with Flag-tagged CD95 ligand (2 ng/ml) and anti-Flag antibody (1 μg/ml). Activation of caspase-8 and caspase-3 was analyzed by western blotting. β-Actin or tubulin served as loading control. The blots are representative for several transfectants analyzed and show caspase activation in clone no. 19 with low expression of c-FLIPR-ΔC and clone no. 3 with high expression of full-length c-FLIPR. (b) For analysis of apoptosis sensitivity, wild-type cells and transfectants were left untreated or stimulated for 16 h with different concentrations of Flag-tagged CD95 ligand (2 and 20 ng/ml) cross-linked with anti-Flag antibody (1 μg/ml). The amount of apoptotic cells was determined by Nicoletti assay. (c) Cells were stimulated with 20 ng/ml CD95 ligand and anti-Flag antibody (1 μg/ml) for 8 and 16 h or left untreated. Caspase-8 activation was analyzed by western blotting as described in (a). Note that the disappearance of the p18 caspase-8 fragment in A20 wild-type cells after CD95 ligand treatment for 16 h is presumably due to massive cell death and the leakage of the p18 fragment into the culture supernatant
As assessed by measurement of DNA fragmentation, expression of full-length c-FLIPR also resulted in an almost complete protection against CD95L-induced apoptosis, whereas the vector control cells were as sensitive as the wild-type cells (Figure 3b). Complete CD95 resistance was observed in several clones with high (clone no. 3; Figure 3b), intermediate and low expression levels of full-length c-FLIPR (data not shown). Interestingly, A20 cells expressing the truncated c-FLIPR-ΔC showed a different behavior that was dependent on the expression level and CD95L concentration. At low CD95L concentrations, cells with high and low levels of c-FLIPR-ΔC expression were similarly protected as full-length c-FLIPR-expressing cells. In contrast, at higher concentrations of CD95L cells with a lower expression of c-FLIPR-ΔC (e.g., clone no. 19) showed some apoptosis induction, even when several clones with comparable expression of the truncated and full-length c-FLIPR proteins were analyzed (compare clone no. 19 and 3; Figures 2b and 3b). This suggests that the C-terminal tail influences the antiapoptotic function of mouse c-FLIPR, which is in contrast to human c-FLIPshort whose C-terminal deletion did not interfere with apoptosis inhibition.20 This conclusion is supported by our observation that incubation of cells with high concentrations of CD95L for longer time periods activated caspase-8 in c-FLIPR-ΔC- but not c-FLIPR-expressing cells, as shown by the appearance of the p18 caspase-8 fragment (Figure 3c).
The C-terminal tail of c-FLIPR contributes to DISC recruitment and antiapoptotic function
It was surprising that despite the increased protein stability (see Figure 2b) c-FLIPR-ΔC was less potent in inhibiting apoptosis when compared with full-length c-FLIPR (see Figure 3b). As c-FLIP proteins act at the receptor level,10, 11 we analyzed DISC formation in the transfected cell lines. To rule out that different expression levels of the transgenes might affect DISC formation, we chose clones with comparable expression of full-length c-FLIPR (clone no. 3) and c-FLIPR-ΔC (clone no. 19). As expected, following a 15-min incubation with CD95L, recruitment of FADD and caspase-8 was observed in wild-type and vector control A20 cells (Figure 4a). In both cell lines, cleavage of caspase-8 to the p43 intermediate was detected, indicating initiation of the death signal. In contrast, caspase-8 processing was undetectable in cells transfected with c-FLIPR-ΔC or full-length c-FLIPR (Figure 4a). Moreover, when the total amount of uncleaved and processed caspase-8 in the DISC was compared, reproducibly less caspase-8 was recruited upon expression of full-length c-FLIPR than in the control cells or FLIPR-ΔC cells (Figure 4a). This is consistent with the observation that c-FLIPR-ΔC was less efficiently recruited into the DISC than the full-length protein (Figure 4a), and could explain the impaired protection at high concentrations of CD95L (see Figure 3b).

Deletion of the C-terminal tail of c-FLIPR reduces DISC recruitment and leads to formation of death effector filaments. (a) Wild-type A20 cells (wt) or cells stably transfected with full-length c-FLIPR, c-FLIPR-ΔC or a Flag-control vector were either stimulated for 15 min with the Flag-tagged CD95 ligand (200 ng/ml) or left untreated. DISC components were co-immunoprecipitated with CD95 using an anti-CD95 antibody (Jo2) coupled to protein-G–sepharose and analyzed by western blotting. (b) NIH/3T3 cells were transiently transfected with either c-FLIPR or c-FLIPR-ΔC. Twenty-four hours post transfection, the cells were fixed and stained with anti-Flag antibody (green) and DAPI (blue). Subsequently, samples were analyzed by confocal laser scanning microscopy
As DISC recruitment of c-FLIP might be influenced by its subcellular localization, we performed immunofluorescence staining of NIH/3T3 cells transiently transfected with either c-FLIPR full-length or c-FLIPR-ΔC. The full-length protein was distributed in the cytosol and partly localized in dotted structures (Figure 4b). c-FLIPR-ΔC was also localized in the cytosol, but showed oligomerization into structures similar to death effector filaments (Figure 4b). Formation of such filaments by c-FLIPR-ΔC but not full-length c-FLIPR was not due to the higher expression of the deletion mutant, as shown in a titration experiment (Supplementary Figure 1). Even at the lowest amount of the transfected DNA, when c-FLIPR-ΔC was not detectable by immunoblotting, filament formation was observed. In contrast, full-length c-FLIPR did not form extensive filaments, although protein expression was much higher. Death effector filaments have been shown to be sites of caspase activation correlating with apoptosis induction.21 Despite formation of these filaments by FLIPR-ΔC, we did not observe apoptosis induction in the transfected cells (data not shown), suggesting that aggregated DEDs of c-FLIP do not build a platform for caspase-8 activation. Nevertheless, retention of c-FLIPR-ΔC in those filaments might explain its reduced DISC recruitment.
Covalently linked tandem DEDs are required for DISC recruitment
Like caspase-8 and caspase-10, but in contrast to FADD, c-FLIP proteins contain two DEDs. Whether the tandem DEDs are simply a duplication of a protein interaction motif or functionally important has not been addressed in vivo. Therefore, we investigated which of the two DEDs of c-FLIPR is required for its antiapoptotic function. To this end, we employed truncation constructs encoding only the first (DED1) or the second DED (DED2) and expressed them stably in A20 cells. Transfectants expressing only DED1 or DED2 showed a marginally reduced sensitivity for CD95-mediated apoptosis (Figure 5a). In addition, no DISC recruitment of the single DEDs and normal caspase-8 activation were observed (Figure 5b). In line, processing of caspase-3 and caspase-8 was comparable to vector control cells (data not shown). Moreover, in contrast to full-length c-FLIPR that was localized in cytoplasmic granular structures, both single DEDs were evenly distributed in the cytosol and nucleus (Figure 5c). Thus, a single DED of c-FLIPR is insufficient to inhibit CD95-mediated apoptosis. We next generated double transfectants expressing both DEDs as separate molecules but without an intramolecular linker, and analyzed their sensitivity toward CD95L. Cells expressing both DEDs were as sensitive as vector control cells (Figure 5a) and showed no DISC recruitment (Figure 5b) and processing of caspase-3 and caspase-8 (data not shown). Thus, an intramolecular linkage seems to be crucial for binding of the tandem DEDs to the DISC. Taken together, both DEDs of mouse c-FLIPR are required for DISC recruitment and its antiapoptotic activity.

DISC recruitment requires intramolecular linkage between the two DEDs. (a) Wild-type A20 cells (wt) or A20 cells stably transfected with c-FLIPR-DED1, c-FLIPR-DED2 or both DEDs were either left untreated or stimulated for 16 h with different concentrations of Flag-tagged CD95 ligand (2 and 20 ng/ml) cross-linked with anti-Flag antibody (1 μg/ml). The amount of apoptotic cells was quantified by Nicoletti assay. (b) Cells expressing Myc-tagged DED1, V5-tagged DED2 or both DEDs were stimulated for 15 min with 200 ng/ml Flag-CD95L or left untreated. DISC components were co-immunoprecipitated with the CD95 receptor using anti-CD95 antibody coupled to protein-G–sepharose and analyzed by western blotting. (c) NIH/3T3 cells were transiently transfected with Flag-FLIPR, DED1-Myc or DED2-V5. Twenty-four hours post transfection, the cells were fixed and stained with tag-specific antibodies, followed by an Alexa-Fluor-conjugated secondary antibody and DAPI. Subsequently, samples were analyzed by confocal laser scanning microscopy
A model of the molecular structure of murine c-FLIPR
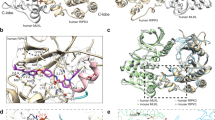
To define the surface residues of murine c-FLIPR involved in interaction with other DISC proteins, we created a structural model of c-FLIPR on the basis of the structure (PDB code, 2BBR) of the v-FLIP MC159.13, 14 The sequence alignment of MC159 and c-FLIPR, and their backbone superposition, are shown in Figure 6a. The homology model of c-FLIPR displays the two helical bundles of the individual DEDs packed against each other in the predicted way. The major difference between c-FLIPR and MC159 is the absence of an additional short α-helix at the beginning of the c-FLIPR DED1, which is due to the slightly shortened N-terminus (data not shown). However, the conspicuous surface motifs, including the charge triad and the hydrophobic patch, which are functionally important for MC159 and caspase-8,13 are very similarly placed within the two structures (Figure 6b, left panel). Interestingly, residues F119, L120 and Y124 are part of an extended hydrophobic patch on the DED2 surface of c-FLIPR, which is not strictly conserved in MC159 (Figure 6b, right panel).

Structural modeling of murine c-FLIPR. (a) Sequence and structural alignment of v-FLIP MC159 and c-FLIPR. The conserved residues in the sequence are marked in bold letters. Color indications are as follows: yellow, conservation of hydrophobic amino acids; blue, conservation of basic amino acids; red, conservation of acidic amino acids and cyan, conservation of small amino acids. Backbone superposition shows that sequence changes of c-FLIPR (blue) relative to v-FLIP (red) are in accordance with relatively small structural differences, resulting in an r.m.s.d. for the backbone atoms of 0.97 Å. (b) Left panel: Representation of hydrophobic and charged motifs within each DED of c-FLIPR. Motifs are colored as follows: charge triad of DED1 in red (E22, R69, D71), hydrophobic patch of DED1 in yellow (M25), charge triad of DED2 in magenta (D113, R166, D168) and hydrophobic patch of DED2 in gold (F119, L120, Y124). For the hydrophobic motif of DED1, only M25 is slightly surface-exposed, whereas the hydrophobic patch containing F119 and L120 is part of a larger shallow hydrophobic cleft on the surface of DED2. Right panel: Hydrophobic amino acids present on the surface of the DED2 hydrophobic patch are highlighted. Hydrophobic regions are marked in yellow and hydrophilic regions are marked in green. The extended hydrophobic surface is composed of the residues F119, L120, L146, L148 and L156. (c) Homology model of murine c-FLIPR. The ribbon structure highlights the secondary structure topology of the two DEDs (DED1 and DED2, colored green and blue, respectively). Similar to v-FLIP, helix 3 of the DED fold is missing, but for reasons of consistency helical numbering is maintained according to the canonical fold and similar to the numbering of the v-FLIP structure 2F1S. The side chains of the residues that were mutated in this study are shown as sticks and are labeled by residue type and number
Our model allowed us to perform mutagenesis based on structural reasoning, and the residues mutated in this study are depicted in Figure 6c. First, we were interested in the role of the hydrophobic patch of DED1 that forms the interface with DED2. We therefore mutated Phe-28 to glycine, since mutation of the corresponding residues in FADD (F25G) and MC159 (F30A) impairs their signaling function.16, 22 The F28G substitution, however, had no effect on caspase processing, DISC formation, subcellular localization or the antiapoptotic function of c-FLIPR (Figure 7). This suggests that the architecture of c-FLIPR was not disturbed and that Phe-28 hydrophobic contacts are not required for stabilization of the DED1/DED2 interface.

The charge triads are not required for DISC binding of c-FLIPR. (a) Different c-FLIPR mutants were generated with mutations either in the charge triad of the first DED (R69A/D71A) or the charge triad of the second DED in c-FLIPR (R166A/D168A). Additionally, the hydrophobic patch (F28G) in DED1 was mutated. The different constructs were stably transfected into A20 cells and apoptosis sensitivity of the transfectants was determined by Nicoletti assay. To this end, cells were either left untreated or stimulated for 16 h with different concentrations of Flag-tagged CD95 ligand (2 and 20 ng/ml) cross-linked with anti-Flag antibody (1 μg/ml). (b) A20 cells transfected with the respective c-FLIPR mutants were stimulated for the indicated time with Flag-tagged CD95 ligand (2 ng/ml) and anti-Flag antibody (1 μg/ml). Cells were lysed and cleavage of caspase-8 and caspase-3 was determined by western blotting. Equal protein loading was checked by probing the blots with anti-β-actin. (c, d) DISC analyses of A20 cells expressing the various c-FLIPR constructs. Cells expressing the DED1 F28G mutant (c), the DED2 charge triad R166A/D168A mutant (c) or the DED1 charge triad R69A/D71A mutant (d) were analyzed in comparison to wild-type A20 cells or cells expressing full-length FLIPR. Cells were either stimulated for 15 min with Flag-tagged CD95 ligand (200 ng/ml) or left untreated. DISC components were co-immunoprecipitated with CD95 using anti-CD95 coupled to protein-G–sepharose and analyzed by western blotting. (e) NIH/3T3 cells were transiently transfected with the indicated c-FLIPR mutants. Twenty-four hours post transfection, cells were stained with anti-Flag-antibody followed by an anti-mouse Alexa-Fluor-488 antibody and DAPI. Subsequently, samples were analyzed under a confocal laser scanning microscope. *An unspecific band recognized by the anti-FLIP antibody
The charge triads of c-FLIPR are dispensable for DISC recruitment
Both DEDs of v-FLIP MC159 contain a hydrogen-bonded cluster of charged amino acids, called the charge triad, which is important for apoptosis inhibition.13, 14, 22 To analyze whether the charge triads play a functional role for cellular FLIP, we mutated both motifs in c-FLIPR (mutants R69A/D71A and R166A/D168A). A20 cells expressing these mutants were not impaired in protection against CD95-mediated apoptosis when compared with c-FLIPR (Figure 7a). Moreover, both charge triad mutants inhibited caspase activation similarly as wild-type c-FLIPR or the F28G mutant (Figure 7b). DISC analysis showed also normal recruitment of the R166A/D168A mutant and no processing of caspase-8 (Figure 7c). The same results were observed with the R69A/D71A mutant (Figure 7d). Thus, in contrast to v-FLIP, the charge triad of c-FLIPR is dispensable for DISC recruitment and its antiapoptotic function.
The hydrophobic patch of DED2 of c-FLIPR is crucial for DISC recruitment
In addition to the charge triad, c-FLIPR contains characteristic hydrophobic epitopes in both DEDs. This hydrophobic patch in DED1 is buried in the DED1/DED2 interface, whereas DED2 contains an extended hydrophobic patch with residues F119, L120, L146, L148 and L154 on its surface (Figure 6b). To investigate the importance of this motif for DISC interaction of c-FLIPR, we mutated Phe-119 and Leu-120 (F119G/L120G) and generated several clones stably expressing this mutant (Figure 8a). Strikingly, all clones showed a complete loss of protection and were as sensitive toward CD95-mediated apoptosis as untransfected cells (Figure 8b). Concomitantly, caspases were readily processed in cells expressing the F119G/L120G upon stimulation with CD95L (Figure 8c). Furthermore, as DISC recruitment of the F119G/L120G mutant was severely diminished, caspase-8 processing could be clearly detected (Figure 8d).

The hydrophobic patch in the DED2 of c-FLIPR is responsible for binding to FADD and caspase-8 in the DISC. (a) A20 transfectants stably expressing an empty pEF-Flag-control vector, full-length c-FLIPR or the c-FLIPR-(F119G/L120G) mutant cloned into the pEF-Flag vector were analyzed by western blotting for the expression of c-FLIPR, CD95, caspase-8 and FADD. Blots were probed with anti-tubulin to control equal protein loading. (b) Apoptosis sensitivity of the different clones was determined by Nicoletti assay. To this end, cells were either left untreated or stimulated for 16 h with different concentrations of Flag-tagged CD95 ligand (2 and 20 ng/ml) cross-linked with anti-Flag antibody (1 μg/ml). (c) A20 cells transfected with the respective c-FLIPR mutants were stimulated for the indicated time with Flag-tagged CD95 ligand (2 ng/ml) and anti-Flag antibody (1 μg/ml). Cells were lysed and cleavage of capase-8 and capase-3 was determined by western blotting. Equal protein loading was checked by probing the blots with anti-β-actin. (d) A20 cells expressing the DED2 hydrophobic patch F119G/L120G mutant (clones no. 12 and 13) were analyzed for DISC formation and compared with wild-type A20 cells or cells expressing full-length FLIPR. Cells were either stimulated for 15 min with Flag-tagged CD95 ligand (200 ng/ml) or left untreated. DISC components were co-immunoprecipitated with CD95 using anti-CD95 coupled to protein-G–sepharose and analyzed by western blotting. (e) NIH/3T3 cells were transiently transfected with the indicated c-FLIPR mutants. Twenty-four hours post transfection, cells were stained with anti-Flag-antibody followed by an anti-mouse Alexa-Fluor-488 antibody and DAPI. Subsequently, samples were analyzed by confocal laser scanning microscopy. *An unspecific band recognized by the anti-FLIP antibody
In agreement with the impaired DISC recruitment, we observed an altered subcellular localization of the protein. The F119G/L120G mutant was not localized in dotted structures or death effector filaments, but evenly distributed in the cytosol (Figure 8e). It is conceivable that not only the interaction of F119G/L120G mutant with DISC components but also its self-oligomerization is affected by mutation of the hydrophobic patch. Similar effects have been observed for mutations in the corresponding hydrophobic patch in caspase-8, which resulted in a loss of FADD binding.13 Thus, the hydrophobic patch of the second DED in mouse c-FLIPR is crucial for DISC recruitment and its antiapoptotic function.
Moreover, unlike MC159, c-FLIPR did not inhibit oligomerization of FADD (Supplementary Figure 2B and C). In fact, cellular FLIP proteins rather enhanced formation of insoluble filaments (Supplementary Figure 2B and C). In summary, our results indicate that, in contrast to v-FLIP MC159, c-FLIP proteins utilize the same interaction motif as caspase-8. While MC159 interferes with FADD self-association, our results imply that c-FLIPR directly competes with caspase-8 for recruitment to the DISC.
Discussion
c-FLIP is expressed in three isoforms arising from differential mRNA splicing. Although human c-FLIPlong and c-FLIPshort are well studied, little is known about the other short splice variant, c-FLIPR, that was initially identified in human Raji B cells17 and later also in some malignant cell lines and primary T cells.8, 10 Here, we report the functional and structural characterization of the novel c-FLIPR isoform in the mouse. Our analysis indicates that c-FLIPR is the only short isoform in the mouse. By modeling the three-dimensional structure of c-FLIPR and by analyzing the mouse DISC in vivo, we characterize for the first time the structural requirements for DISC recruitment of cellular FLIP proteins. Our results suggest that mouse c-FLIPR and v-FLIP MC159 inhibit death receptor-mediated apoptosis by different mechanisms.
The unique C-terminal tail and covalently linked tandem DEDs are important for DISC recruitment of c-FLIPR
To determine the structural requirements of c-FLIP proteins for DISC recruitment, we generated several deletion and point mutants and found that a linkage between both DEDs of c-FLIPR is required for its antiapoptotic function. Neither DED1 nor DED2 was recruited into the CD95 DISC, which is presumably caused by the compact structure of the tandem DED (Figure 6),13, 14 and is also consistent with the incapacity of the single DEDs of viral MC159 to protect cells against apoptosis.23
We also analyzed the function of the C-terminal tail of c-FLIPR, which distinguishes the protein from the other two isoforms. Surprisingly, at comparable expression levels c-FLIPR-ΔC less efficiently protected against apoptosis and displayed an impaired DISC recruitment when compared with the full-length protein. The reduced antiapoptotic activity of c-FLIPR-ΔC is in contrast to C-terminal deletion mutants of human c-FLIPshort or MC159, which suppressed death receptor-mediated apoptosis equally as the wild-type proteins.20, 23 Accordingly, an equal incorporation of c-FLIPshort and its C-terminal deletion mutant into the TRAIL-DISC has been observed.20 Although we cannot exclude that the disparity of the mutants in DISC binding might be attributed to different experimental systems, our results suggest that the C-terminus of c-FLIPR mediates DISC interaction. A contribution of the C-terminus to DISC binding is therefore the first described functional difference between c-FLIPR and c-FLIPshort. In addition, we show that mouse c-FLIPR is rapidly degraded and that the C-terminal tail determines protein stability. Since mutation of two lysines (K196, K200), however, did not prevent ubiquitination, the C-terminus is presumably not the direct site of ubiquitination, but seems to regulate protein stability indirectly, for example, by composing a binding site for an E3 ubiquitin ligase. The use of different target residues for ubiquitin conjugation therefore constitutes an additional difference between c-FLIPshort and c-FLIPR. Thus, the C-terminal tail appears to regulate not only DISC recruitment but also degradation of c-FLIPR via the proteasome pathway.
The hydrophobic patch of DED2 is indispensable for c-FLIP function
The three-dimensional structures of MC159 and FADD revealed various characteristic DED motifs, including a charge triad and hydrophobic patch.13, 14, 15 The former includes Arg-69 and Asp-71 in DED1, as well as Arg-166 and Asp-168 in DED2 of c-FLIPR (Figure 6b). On the basis of structural data, this motif has been proposed to be important for protein–protein interactions of DED-containing proteins.14 In contrast to MC159, in which mutations in the charge triad motifs of DED1 or DED2, abolished its antiapoptotic potential,13 the charge triads of c-FLIPR are not absolutely required for DISC binding and apoptosis inhibition (Figure 7). We observed a slightly impaired DISC recruitment of c-FLIPR mutated in either of the two charge triads, which might be explained by the fact that the charge triad participates in stabilization of the DED fold.15 Indeed, our model shows that R166 and D168 are involved in a hydrogen bond network stabilizing contacts to helix I (backbone carbonyl of S108) and helix II (D113) of DED2. Nevertheless, the antiapoptotic function of the R166A/D168A and the R69A/D71A mutants was fully preserved, arguing that the charge triads are not crucial for cellular FLIPs.
In contrast to the mutants discussed above, mutation of the hydrophobic patch in DED2 (F119G/L120G) completely abolished DISC recruitment and the antiapoptotic function of c-FLIPR (Figure 8). The F119G/L120G mutant showed a different localization similar to the single DED mutants that were also not recruited into the DISC. Thus, next to a covalent linkage of the tandem DEDs, the hydrophobic patch of DED2 is crucial for DISC recruitment and the function of c-FLIPR. Importantly, also mutation of the hydrophobic patch in DED2 of caspase-8 has been shown to abolish its interaction with FADD, whereas the corresponding mutation in v-FLIP MC159 had no effect.13
The distinct effects of mutation of the surface motifs indicate that viral and cellular FLIPs employ different mechanisms to inhibit apoptosis. MC159 has been suggested to block apoptosis by inhibiting the oligomerization of FADD, thereby impairing its interaction with death receptors.18 As c-FLIPR and caspase-8 use the same motif for FADD interaction (Figure 8; Rasper et al.18), it is likely that the two molecules show competitive binding to the adapter protein. To investigate these possibilities, we expressed caspase-8 and FADD together with c-FLIPR or MC159. The known insolubility and assembly of the DED proteins into death effector filaments did not allow us to directly show a competitive binding c-FLIPR and caspase-8 to the FADD protein. Nevertheless, consistent with its inhibitory effect on FADD oligomerization, we found that MC159 also inhibited the formation of death filaments (Supplementary Figure 2). c-FLIPR, in contrast, enhanced the assembly of FADD into death effector filaments. Although these distinct effects on FADD oligomerization in death effector filaments might not necessarily reflect their physiological function, it further supports the view that viral and cellular FLIPs employ different mechanisms to inhibit death receptor-mediated apoptosis.
Materials and Methods
Cell culture and transfections
The mouse B-cell line A20 was cultured in RPMI 1640 (PAA Laboratories, Cölbe, Germany) supplemented with 10% fetal calf serum (BioWest, Frickenhausen, Germany), 2 mM glutamine, 50 μM β-mercaptoethanol and 50 μg/ml of each penicillin and streptomycin (Invitrogen, Karlsruhe, Germany). NIH/3T3 cells and 293T human embryonic kidney cells were cultured in Dulbecco's modified Eagle's medium (DMEM high glucose; PAA Laboratories) supplemented with 10% fetal calf serum and 50 μg/ml of each penicillin and streptomycin. Transient transfections of 293T cells and NIH/3T3 cells were performed with FUGENE 6 (Roche, Mannheim, Germany) and Optifect (Invitrogen) reagent according to the manufacturers' protocols. For stable transfection, 1 × 107 A20 cells were pulsed with 10 μg DNA at 100 V and 750 μF (Gene Pulser Xcell; Bio-Rad, Munich, Germany). Stably transfected clones were selected by limited dilution in medium containing 4 μg/ml puromycin (Sigma, Deisenhofen, Germany), 500 μg/ml blasticidin (Invitrogen) or 1 mg/ml G418 (PAA Laboratories). Transgene expression was analyzed by Western blotting. The constructs for GFP-FADD21 and MC1597 have been described earlier.
Cloning of murine c-FLIPR and mutagenesis
Total RNA was isolated from the thymus of a C57Bl/6 mouse by acidic phenol–guanidinium thiocyanate–chloroform extraction.24 RNA was reverse transcribed with Superscript III (Invitrogen) and PCR was performed with Pfx proof-reading polymerase (Invitrogen). The primers used were designed according to the published murine CASHβ sequence: 5′-GGATGGAGACTGGACGAGAAC-3′ (forward primer) and 5′-CCACAGTAGTCATGCCCAGAT-3′ (reverse primer). c-FLIPR mutants were generated by using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) and verified by DNA sequencing. Primer sequences are available upon request. Wild-type c-FLIPR, c-FLIPR-ΔC and all point mutants were cloned into pEF-FLAG.10 DED1 was cloned in frame with the Myc-tag of pEF/Myc/cyto (Invitrogen) and DED2 in frame with the V5-His-tag of pEF6/V5-His-TOPO (Invitrogen).
FACS assays
Cell surface staining and cytotoxicity assays were performed as described previously.19 For assaying apoptosis, 1 × 106 cells were stimulated in 24-well plates (if not stated otherwise) with 2 or 20 ng/ml CD95L and anti-Flag (M2; 1 μg/ml; Sigma), or left untreated for the indicated time periods.
Western blot analysis
For western blot analysis, cells were lysed in TPNE buffer (PBS adjusted to 300 mM NaCl, 1% Triton X-100, 2 mM EDTA, 1 mM PMSF and 1 μg/ml each of leupeptin, aprotinin, chymostatin and pepstatin A). A 20-μg weight of post-nuclear supernatant protein, as determined by the BCA method (Pierce Biotechnology, Rockford, IL, USA), was separated by 12% SDS-PAGE, blotted onto a nitrocellulose membrane (Amersham, Freiburg, Germany) and blocked with 5% non-fat dry milk in PBS/Tween (0.05% Tween-20 in PBS). After washing with PBS/Tween, the blots were incubated overnight with specific antibodies at 4°C. Blots were washed again with PBS/Tween, incubated with horseradish peroxidase (HRPO)-coupled secondary antibodies (1:20 000) for 1 h at room temperature, washed again and developed with a chemiluminescence reagent (Amersham). For stripping, blots were incubated in Re-Blot mild solution (Chemikon, Hofheim, Germany) according to the manufacturer's procedure.
The antibodies used for western blotting and immunoprecipitation were as follows: β-actin (AC-74; Sigma), caspase-3 (46; BD Biosciences, Heidelberg, Germany), caspase-8 (1G12; Alexis, Gruenberg, Germany), FADD (1F7; Upstate, Hamburg, Germany), CD95 (Jo2; BD Biosciences), CD95 (M20; Santa Cruz Biotechnology, Heidelberg, Germany), Flag (M2; Sigma), c-MYC (9E10; Santa Cruz Biotechnology), V5 (Invitrogen) and tubulin (Molecular Probes, Goettingen). HRPO-conjugated goat anti-rabbit IgG was from Santa Cruz Biotechnology. HRPO-conjugated goat anti-rat IgG, donkey anti-goat IgG, goat anti-mouse IgG1, IgG2a and IgG2b were from Southern Biotechnology Associates (Birmingham, AL, USA).
Immunoprecipitations
For DISC analysis, 2 × 107 A20 cells were stimulated with 200 ng/ml Flag-CD95L or left untreated. If not stated otherwise, stimulation was stopped with cold PBS after 15 min. Cells were lysed in DISC buffer (20 mM Tris/HCl, pH 7.4, 1% Triton X-100, 10% glycerol, 150 mM NaCl, 1 mM PMSF and 1 μg/ml of leupeptin, aprotinin, chymostatin and pepstatin A) for 15 min on ice and centrifuged (15 min, 14 000 × g). Subsequently, the DISC was precipitated with 2 μg anti-CD95 (Jo2) coupled to protein G beads (Sigma) for 4 h at 4°C. Finally, the beads were washed three times with 750 μl ice-cold DISC buffer and analyzed by western blotting.
To analyze ubiquitination of c-FLIPR, HEK293T cells were transiently transfected with His–ubiquitin with or without c-FLIPR full-length or c-FLIPR-ΔC using FUGENE 6 transfection reagent. 20 h later, cells were incubated with 10 μg/ml MG132 for further 4 h. Then, cells were washed and lysed in buffer A (6 M urea, 0.1 M sodium phosphate, 10 mM Tris/HCl pH 8, 5 mM imidazole, 10 mM β-mercaptoethanol). Lysates were sonicated and incubated with 20 μl Ni-NTA agarose (Qiagen) for 4 h at room temperature. Beads were washed with 1 ml each of buffer A, buffer B (6 M urea, 0.1 M sodium phosphate, 10 mM Tris/HCl pH 8, 10 mM β-mercaptoethanol) and buffer C (6 M urea, 0.1 M sodium phosphate, 10 mM Tris/HCl pH 6.3, 10 mM β-mercaptoethanol). Subsequently, precipitated proteins were analyzed by western blotting.
Fluorescence microscopy
A total of 4 × 104 NIH/3T3 cells grown on a coverslip in a 12-well dish were transiently transfected with 4 μg plasmid DNA using Optifect transfection reagent. Twenty-four hours later, cells were washed with PBS and fixed with 3.7% paraformaldehyde in PBS for 15 min. The cells were washed again and permeabilized with 0.05% saponin and 4% BSA in PBS for 30 min. Thereafter, cells were incubated with the anti-Flag antibody overnight in blocking solution (4% BSA, 0.05% saponin in PBS) at 4°C. Coverslips were washed several times with PBS and incubated with anti-mouse Alexa-Fluor-488 secondary antibody (Molecular Probes) for 1 h at room temperature. For nuclear staining, the cells were washed several times and incubated with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI, 100 ng/ml) for 10 min. Cells were then mounted in mounting medium (DakoCytomation, Hamburg, Germany) and analyzed under a confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany) at a magnification of × 630.
Structural modeling
Structural modeling was based on the 1.2-Å resolution crystal structure 2BBR of MC159.13 According to the sequence alignment presented in Figure 6a, the residues 4–17, 21–38, 42–88 and 92–188 of 2BBR were mutated by the equivalent amino-acid side chains preserving positions of backbone atoms, using the biopolymer module of Sybyl7.2.5 (Tripos Inc., St Louis, MO, USA). The gaps were modeled by means of the loop search procedure of Sybyl7.2.5. To relax the non-conserved residues of the c-FLIPR-model, the structure was minimized using harmonic restraints for all conserved residues (Amber8, 10 000 steps). Finally, the model was minimized again without any restraints (Amber8, 30 000 steps). The root mean-square distance (r.m.s.d.) of the heavy backbone atoms between 2BRR and the c-FLIPR-model is 0.97.
Abbreviations
- CD95L:
-
CD95 ligand
- c-FLIP:
-
cellular FLICE-inhibitory protein
- CHX:
-
cycloheximide
- DED:
-
death effector domain
- DISC:
-
death-inducing signaling complex
- FADD:
-
Fas-associated death domain
- v-FLIP:
-
viral FLICE-inhibitory protein
References
Peter ME, Krammer PH . The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 2003; 10: 26–35.
Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM et al. A unified model for apical caspase activation. Mol Cell 2003; 11: 529–541.
Chang DW, Xing Z, Capacio VL, Peter ME, Yang X . Interdimer processing mechanism of procaspase-8 activation. EMBO J 2003; 22: 4132–4142.
Donepudi M, Mac Sweeney A, Briand C, Grutter MG . Insights into the regulatory mechanism for caspase-8 activation. Mol Cell 2003; 11: 543–549.
Krueger A, Baumann S, Krammer PH, Kirchhoff S . FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol 2001; 21: 8247–8254.
Budd RC, Yeh WC, Tschopp J . cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol 2006; 6: 196–204.
Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997; 386: 517–521.
Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN . c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem 2005; 280: 14507–14513.
Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388: 190–195.
Scaffidi C, Schmitz I, Krammer PH, Peter ME . The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 1999; 274: 1541–1548.
Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S . Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem 2001; 276: 20633–20640.
Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H . The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol 2007; 25: 561–586.
Yang JK, Wang L, Zheng L, Wan F, Ahmed M, Lenardo MJ et al. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell 2005; 20: 939–949.
Li FY, Jeffrey PD, Yu JW, Shi Y . Crystal structure of a viral FLIP: insights into FLIP-mediated inhibition of death receptor signaling. J Biol Chem 2006; 281: 2960–2968.
Carrington PE, Sandu C, Wei Y, Hill JM, Morisawa G, Huang T et al. The structure of FADD and its mode of interaction with procaspase-8. Mol Cell 2006; 22: 599–610.
Eberstadt M, Huang B, Chen Z, Meadows RP, Ng SC, Zheng L et al. NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature 1998; 392: 941–945.
Djerbi M, Darreh-Shori T, Zhivotovsky B, Grandien A . Characterization of the human FLICE-inhibitory protein locus and comparison of the antiapoptotic activity of four different flip isoforms. Scand J Immunol 2001; 54: 180–189.
Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SL et al. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ 1998; 5: 271–288.
Schmitz I, Weyd H, Krueger A, Baumann S, Fas SC, Krammer PH et al. Resistance of short term activated T cells to CD95-mediated apoptosis correlates with de novo protein synthesis of c-FLIPshort. J Immunol 2004; 172: 2194–2200.
Poukkula M, Kaunisto A, Hietakangas V, Denessiouk K, Katajamaki T, Johnson MS et al. Rapid turnover of c-FLIPshort is determined by its unique C-terminal tail. J Biol Chem 2005; 280: 27345–27355.
Siegel RM, Martin DA, Zheng L, Ng SY, Bertin J, Cohen J et al. Death-effector filaments: novel cytoplasmic structures that recruit caspases and trigger apoptosis. J Cell Biol 1998; 141: 1243–1253.
Garvey TL, Bertin J, Siegel RM, Wang GH, Lenardo MJ, Cohen JI . Binding of FADD and caspase-8 to Molluscum contagiosum virus MC159 v-FLIP is not sufficient for its antiapoptotic function. J Virol 2002; 76: 697–706.
Garvey T, Bertin J, Siegel R, Lenardo M, Cohen J . The death effector domains (DEDs) of the Molluscum contagiosum virus MC159 v-FLIP protein are not functionally interchangeable with each other or with the DEDs of caspase-8. Virology 2002; 300: 217–225.
Sambrook J, Russel DW . Molecular Cloning – A Laboratory Manual. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, 2001.
Acknowledgements
We thank Carina Meyer and Daniel Scholtyssik for expert technical assistance, and Dr Christian Schwerk for helpful discussions. We are also grateful to Drs Vishva Dixit, Thomas Hofmann, Michael Lenardo, Margot Thome and Harald Wajant for various reagents. This work was supported by grants from the Forschungskommission, Faculty of Medicine Düsseldorf, by the Deutsche Krebshilfe and the Deutsche Forschungsgemeinschaft (GK1033).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by A Ashkenazi
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Ueffing, N., Keil, E., Freund, C. et al. Mutational analyses of c-FLIPR, the only murine short FLIP isoform, reveal requirements for DISC recruitment. Cell Death Differ 15, 773–782 (2008). https://doi.org/10.1038/sj.cdd.4402314
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402314
Keywords
This article is cited by
-
c-FLIP regulates autophagy by interacting with Beclin-1 and influencing its stability
Cell Death & Disease (2021)
-
c-FLIP is crucial for IL-7/IL-15-dependent NKp46+ ILC development and protection from intestinal inflammation in mice
Nature Communications (2020)
-
Long and short isoforms of c-FLIP act as control checkpoints of DED filament assembly
Oncogene (2020)
-
c-FLIP and CD95 signaling are essential for survival of renal cell carcinoma
Cell Death & Disease (2019)
-
Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain
Cell Death & Differentiation (2016)
